Effect of defects on photocatalytic dissociation of methanol on TiO2(110)†
Chuanyao
Zhou‡
,
Zhibo
Ma‡
,
Zefeng
Ren
*,
Xinchun
Mao
,
Dongxu
Dai
and
Xueming
Yang
*
State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, 457 Zhongshan Road, Dalian, 116023, Liaoning, P. R. China. E-mail: zfren@fhi-berlin.mpg.de; xmyang@dicp.ac.cn
First published on 22nd July 2011
Abstract
Photocatalytic dissociation of deuterated methanol (CD3OD) on both stoichiometric and reduced TiO2(110) surfaces was investigated using the time-dependent two-photon photoemission (2PPE) method, in order to understand the effect of defects on the kinetics of methanol dissociation on TiO2(110). By monitoring the time evolution of the photoinduced excited state on the methanol covered surface, the photocatalytic dissociation kinetics of methanol on the TiO2 surface were observed. The measured photodissociation rate on the reduced TiO2(110) surface is more than an order of magnitude faster than that on the stoichiometric surface. Since the reduced TiO2(110) surface has considerably more surface and subsurface defects than the stoichiometric surface, the experimental observation suggests that one or both of them could accelerate the photocatalysis process of methanol on the TiO2(110) surface in a significant way.
1) Introduction
Simple alcohols are often used as model systems of catalytic and photocatalytic oxidation of organic contaminants on TiO21–3 and as a probe to the active sites on metal oxides and related materials.4–8 Among them, methanol on TiO2 has attracted much attention in both experimental9–12 and theoretical13–17 investigations because of the remarkable enhancement effect in photocatalyzed hydrogen production in the mixture of methanol with water.18 Dissociation of methanol on bridge-bonded oxygen vacancy sites (BBOv's)19 of TiO2(110) surfaces has been observed by scanning tunneling microscopy (STM), and this suggests that spontaneous dissociation at these oxygen vacancy sites is possible, in accordance with the recent results of theoretical calculations.14 However, the role of the in-plane Ti5c sites, on which methanol can also be adsorbed molecularly, in the TiO2 catalysis or photocatalysis has not been clarified until recently. The methanol/TiO2(110) surface has been studied previously using the two-photon photoemission (2PPE) method,11,12 and an unoccupied excited surface state was observed at about 2.4 eV above the Fermi level (EF). In a more recent study using the time-dependent two-photon photoemission (TD-2PPE) technique, in combination with STM study and DFT calculations, we have found that the unoccupied excited surface state on the methanol/TiO2(110) surface is photoinduced and attributed this state to the photocatalyzed dissociation of methanol on TiO2(110).20 More interestingly, the photocatalyzed dissociation of methanol on TiO2(110) seems to occur mainly on the Ti5c sites, not on the BBOv's. Previously, surface defect sites were often regarded as the active reaction sites for catalysis and photocatalysis. Therefore it is quite surprising that oxygen defects are not the active centers for photocatalysis of methanol on TiO2. This, however, does not mean that oxygen defects do not have any effect on the photocatalytic process. In addition, subsurface defects such as Ti interstitials and subsurface oxygen vacancies can also affect the surface chemistry on TiO2.21–26 In order to investigate the specific role of the defects in the photocatalytic chemistry of methanol on TiO2(110), we have designed a comparative experimental study of photochemistry of methanol on both stoichiometirc (with few defects) and reduced (more defects) TiO2(110) surfaces using the TD-2PPE technique.20,27 In this work, photodissociation rates of CD3OD (1ML) on both stoichiometric and reduced TiO2(110) surfaces were measured quantitatively and also directly compared. The experimental results clearly show that the defects on and below the TiO2(110) surface quite remarkably affect the kinetics of photocatalytic methanol dissociation on the TiO2 surface by lowering the reaction barrier by a considerable amount.2) Experimental
The current experiments were carried out using a newly developed apparatus that has the capability of measuring the TD-2PPE signal for the methanol covered TiO2(110) surface.27 The instrument consists of an ultrahigh vacuum chamber (base pressure 5 × 10−11 mbar) and a Ti:Sapphire femtosecond laser oscillator system (Femtolasers, 75 MHz). The reduced TiO2(110) surface was prepared by cycles of Ar+ sputtering and UHV annealing at 850 K, whereas the stoichiometric surface was prepared by repeated cycles of Ar+ sputtering and annealing at 850 K in a low oxygen background (1 × 10−7 mbar) followed by cooling in the same condition to 450 K. According to the temperature programmed desorption (TPD) of the water method, the reduced surface has an oxygen defect density of about 6%,28 while the stoichiometric surface has nearly no oxygen defect.29 Furthermore, the density of subsurface defects on reduced TiO2(110) is also larger,21,22,30 though quantitative assessment of them is not possible in the current experiment. As annealing in the oxygen background to prepare a TiO2(110) with little defects will generate a roughened TiO2(110) surface31 which may affect the surface chemistry of methanol. We therefore prepared a nearly perfect surface using a method developed by Besenbacher and coworkers22 for comparison studies. The CD3OD sample was purchased from Cambridge Isotope Laboratories, and was further purified by several freeze-pump-thaw cycles. A monolayer (ML) of CD3OD on TiO2(110) surface was prepared by over dosing the fully deuterated methanol sample on the surface at 110 K followed by heating to 190 K according to the TPD spectra of methanol on TiO2(110)10 to get rid of the extra methanol overlayers. The methanol monolayer covered surface was then re-cooled to 110 K for TD-2PPE measurements. A more detailed description of the TD-2PPE experimental procedures can be found in the supplementary information. By using the TD-2PPE technique, the rate of photocatalyzed methanol dissociation on TiO2 can be measured quantitatively. This provides us a good chance to investigate the effect of defects on the photocatalytic chemistry of this prototype system.3) Results and discussion
Fig. 1 shows the TD-2PPE spectra for 1 ML CD3OD covered stoichiometric TiO2(110) (s-TiO2(110)) and reduced TiO2(110) (r-TiO2(110)) surfaces as a function of laser irradiation time. Both 1 ML CD3OD covered stoichiometric and reduced TiO2(110) surface were illuminated by a femtosecond laser beam with exactly the same center wavelength and power (401.6 nm, 70.4 mW). On both surfaces, a similar photoinduced excited electronic resonance between 5 eV and 6 eV was observed, which has been previously assigned to the photocatalyzed methanol dissociation state on the Ti5c sites.20 The rise times for the excited resonance, however, are obviously not the same for the two surfaces.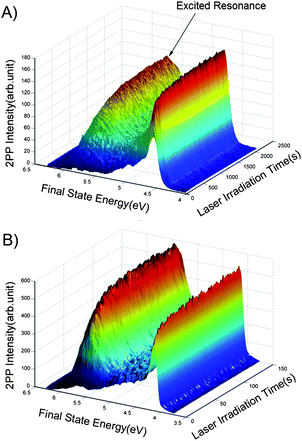 | ||
| Fig. 1 2PPE spectra for the 1 ML CD3OD covered stoichiometric (A) and reduced (B) TiO2(110) surfaces as a function of the laser irradiation time. The center wavelength and power of the laser were exactly the same for the two experimental measurements. The energy reference is the Fermi level. | ||
In order to determine the rate of photocatalyzed methanol dissociation on both stoichiometric and reduced surfaces, we have integrated the excited state signal between 4.9 eV and 6.1 eV in Fig. 1, and obtained the total excited resonance signal as a function of laser irradiation time (see Fig. 2) for both surfaces. As discussed in our previous work,20 the kinetics of an excited state rising cannot be described by a single exponential function. This shows that the kinetics of photocatalyzed methanol dissociation is of a heterogeneous nature. The time dependent excited resonance signal of the 2PPE spectra can be simulated very well by the fractal kinetic model:32
| I = I0 (1 − exp(−k0/(1 − h) t(1−h))) | (1) |
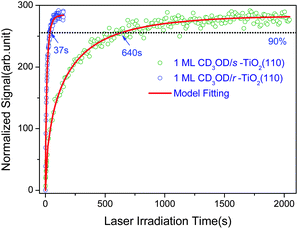 | ||
| Fig. 2 Normalized time dependent signal of the excited resonance of 1 ML CD3OD covered stoichiometric (olive circles) and reduced (blue circle) TiO2(110) surface and the fractal kinetics model fittings (red lines). | ||
The k0 constant and h parameter obtained for methanol photocatalytic dissociation at both stoichiometric and reduced TiO2 surfaces are listed in Table 1. The physical meaning of the fractal kinetics is, however, not immediately clear. According to recent results using mass spectrometric studies, the photocatalytic dissociation of methanol on TiO2(110) seems to go through multiple steps, which could cause fractal like kinetics. The transport of photoexcited electrons and/or holes through a percolation phenomena might be also responsible for the fractal kinetics of the photocatalyzed dissociation of methanol on TiO2(110). More detailed investigations are needed in order to reveal the nature of this interesting kinetics.
| TiO2 Surface | k 0 | h |
|---|---|---|
| Stoichiometric | 0.028(0) | 0.39(0) |
| Reduced | 0.110(2) | 0.24(1) |
A rather dramatic difference between the photocatalyzed methanol dissociation rates on the two surfaces has been observed. The dissociation rate at t = 1 s on the reduced surface is about 4 times of that on the stoichiometeric surface. One can see from Fig. 2, it took only 37 s for the excited resonance signal on the reduced surface to rise to 90% of the maximum signal level, while for the stoichiometric surface, it took 640 s. This is a difference of 17.3 times in the rates.
As pointed out above, using annealing in the oxygen background to prepare a TiO2(110) surface with little defects could cause surface roughening. We therefore used another approach22 to prepare a nearly perfect surface for comparative photochemistry studies. The reduced TiO2(110) with about 6% of BBOv's was exposed to 5 L O2 at 120 K and then annealed in UHV at 550 K for 2 mins. Only a few BBOv's and TiOx islands were left on this surface using this procedure. The photocatalytic dissociation rate was measured on this TiO2(110) surface in comparison with a surface that had about 6% BBOv's. The excited resonance signal on the nearly perfect surface reached 90% of its maximum in 570 s while on the reduced surface, it took only 52 s. Therefore, the photocatalytic dissociation rate on the reduced surface is about 11 times faster (see Fig. S4 in the supplementary information†), which is slightly smaller than the result (17.2 times) shown above (the excitation laser power in these two sets of experiments is different). This is partly due to the measurement errors as the excitation laser power does not change the difference between the photocatalyzed dissociation rate of methanol on stoichiometric and reduced TiO2(110) surfaces (data not shown). The result of this experiment shows that surface roughening could have some small effect on the photochemical rate, but the major difference of the photochemical rates is obviously related to the difference of defect density.
There are surface defects (oxygen vacancies) and subsurface defects (Ti interstitials, oxygen defects) on the TiO2 sample. It has been shown that surface defects33,34 and also the subsurface defects21–26 could have a remarkable effect on chemical reactivity on TiO2(110). Therefore, we believe that the large enhancement of the photocatalytic methanol dissociation rate on the reduced TiO2 surface should be mainly caused by the large density of the defects on the surface. This implies that surface defects and/or subsurface defects could accelerate the photocatalytic methanol dissociation on TiO2 in a remarkable way. However, as the surface defects and subsurface defects affect the surface chemistry on TiO2 similarly,23,24 experimental characterization of their relative contribution is difficult. The acceleration of the photocatalytic methanol dissociation on the TiO2(110) surface seems to originate from the decrease of the reaction barrier caused by the increase of the density of defects (Fig. 3).
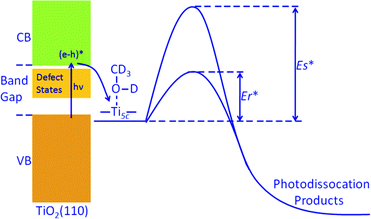 | ||
| Fig. 3 Schematic energy diagram of photodissociation of methanol at Ti5c sites of TiO2(110). Er* stands for the photocatalytic dissociation barrier for the reduced TiO2(110) surface, while Es* is the barrier for the stoichiometric surface. | ||
4) Conclusions
In summary, we have investigated the photochemistry of methanol on both stoichiometric and reduced TiO2(110) surfaces using the time-dependent 2PPE technique. Even though photocatalyzed dissociation of methanol occurs mainly on the Ti5c sites on TiO2(110), defects seem to have a remarkable effect on the photocatalytic kinetic process. Experimental results in this work clearly show that surface defects and/or subsurface defects on the TiO2(110) surface could enhance the photocatalyzed methanol dissociation rate by significantly lowering the photoreaction barrier.Acknowledgements
This work was supported by the Chinese Academy of Sciences, National Natural Science Foundation of China and the Ministry of Science and Technology. We appreciate the insightful discussions with Dr Hongjun Fan. Helpful comments by Prof. Dr Alec Wodtke are also much appreciated. We are also grateful to Qing Guo, Chenbiao Xu and Wenshao Yang for measuring the concentration of BBOv's of the reduced TiO2(110).Notes and references
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- F. Guzman and S. S. C. Chuang, J. Am. Chem. Soc., 2010, 132, 1502–1503 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328–2353 RSC.
- Z. Dohnalek, I. Lyubinetsky and R. Rousseau, Prog. Surf. Sci., 2010, 85, 161–205 CrossRef CAS.
- S. Rousseau, O. Marie, P. Bazin, M. Daturi, S. Verdier and V. Harle, J. Am. Chem. Soc., 2010, 132, 10832–10841 CrossRef CAS.
- L. Nakka, J. E. Molinari and I. E. Wachs, J. Am. Chem. Soc., 2009, 131, 15544–15554 CrossRef CAS.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Surf. Sci., 1998, 413, 252–272 CrossRef.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Faraday Discuss., 1999, 114, 313–329 RSC.
- K. Onda, B. Li, J. Zhao and H. Petek, Surf. Sci., 2005, 593, 32–37 CrossRef CAS.
- B. Li, J. Zhao, K. Onda, K. D. Jordan, J. L. Yang and H. Petek, Science, 2006, 311, 1436–1440 CrossRef CAS.
- S. P. Bates, G. Kresse and M. J. Gillan, Surf. Sci., 1998, 409, 336–349 CrossRef CAS.
- R. S. de Armas, J. Oviedo, M. A. San Miguel and J. F. Sanz, J. Phys. Chem. C, 2007, 111, 10023–10028 Search PubMed.
- J. Oviedo, R. Sanchez-De-Armas, M. A. S. Miguel and J. F. Sanz, J. Phys. Chem. C, 2008, 112, 17737–17740 CAS.
- J. Zhao, J. L. Yang and H. Petek, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235416–235426 CrossRef.
- V. M. Sanchez, J. A. Cojulun and D. A. Scherlis, J. Phys. Chem. C, 2010, 114, 11522–11526 CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694–695 RSC.
- Z. R. Zhang, O. Bondarchuk, J. M. White, B. D. Kay and Z. Dohnalek, J. Am. Chem. Soc., 2006, 128, 4198–4199 CrossRef CAS.
- C. Y. Zhou, Z. F. Ren, S. J. Tan, Z. B. Ma, X. C. Mao, D. X. Dai, H. J. Fan, X. M. Yang, J. LaRue, R. Cooper, A. M. Wodtke, Z. Wang, Z. Y. Li, B. Wang, J. L. Yang and J. G. Hou, Chem. Sci., 2010, 1, 575–580 RSC.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. S. Li, J. O. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Laegsgaard, B. Hammer and F. Besenbacher, Science, 2008, 320, 1755–1759 CrossRef CAS.
- E. Lira, J. O. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Laegsgaard, B. Hammer, S. Wendt and F. Besenbacher, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- N. A. Deskins, R. Rousseau and M. Dupuis, J. Phys. Chem. C, 2010, 114, 5891–5897 CAS.
- J. Haubrich, E. Kaxiras and C. M. Friend, Chem.–Eur. J., 2011, 17, 4496–4506 CrossRef CAS.
- U. Aschauer, Y. B. He, H. Z. Cheng, S. C. Li, U. Diebold and A. Selloni, J. Phys. Chem. C, 2010, 114, 1278–1284 CAS.
- C. M. Friend, L. Benz, J. Haubrich and S. C. Jensen, ACS Nano, 2011, 5, 834–843 CrossRef.
- Z. F. Ren, C. Y. Zhou, Z. B. Ma, C. L. Xiao, X. C. Mao, D. X. Dai, J. LaRue, R. Cooper, A. M. Wodtke and X. M. Yang, Chin. J. Chem. Phys., 2010, 23, 255–261 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- K. Onda, B. Li and H. Petek, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045415–045126 CrossRef.
- M. A. Henderson, Surf. Sci., 1995, 343, L1156–L1160 CrossRef CAS.
- M. Li, W. Hebenstreit, L. Gross, U. Diebold, M. A. Henderson, D. R. Jennison, P. A. Schultz and M. P. Sears, Surf. Sci., 1999, 437, 173–190 CrossRef CAS.
- R. Kopelman, Science, 1988, 241, 1620–1626 CAS.
- N. G. Petrik, Z. R. Zhang, Y. G. Du, Z. Dohnalek, I. Lyubinetsky and G. A. Kimmel, J. Phys. Chem. C, 2009, 113, 12407–12411 CAS.
- S. Tan, Y. Ji, Y. Zhao, A. Zhao, B. Wang, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2011, 133, 2002–2009 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00249j |
| ‡ Who have made similar contributions to this work. |
| This journal is © The Royal Society of Chemistry 2011 |