Au@organosilica multifunctional nanoparticles for the multimodal imaging†
Yan
Cui
a,
Xiao-Shan
Zheng
a,
Bin
Ren
*a,
Rui
Wang
b,
Jun
Zhang
b,
Ning-Shao
Xia
b and
Zhong-Qun
Tian
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, CNRS Laboratoire International Associé XiamENS, Key Laboratory of Analytical Sciences, and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: bren@xmu.edu.cn; Fax: +86 592-2181906; Tel: +86 592-2186532
bNational Institute of Diagnostics and Vaccine Development in Infectious Disease, School of Life Sciences, Xiamen University, Xiamen, 361005, China
First published on 23rd June 2011
Abstract
The increasing application of nanomaterials in biosensor and imaging sets a higher demand on the multifunctionalities of nanomaterials for obtaining multiple parameters of a same system under a same condition. In this work, multifunctional Au@organosilica nanoparticles with high stability were conveniently synthesized by direct hydrolyzing of 3-mercaptopropyltriethoxysilane in an aqueous solution in the presence of a Au core. Modification of the Au core with Raman reporters and the organosilica shell with fluorophore before and after the hydrolysis, respectively, produces multifunctional nanoparticles exhibiting Rayleigh scattering of the Au core, fluorescence signals of the fluorophores and surface-enhanced Raman scattering (SERS) of the Raman reporters. The nanoparticles can be used as multimodal tracers for living cell imaging and related biological research.
Introduction
The use of nanomaterials for bio-related imaging and assay has received increasing interest in recent years. With their unique electronic, optical, and magnetic properties, nanomaterials have found wide applications in areas such as biosensors, probe detection, drug delivery, multimodal imaging, simultaneous diagnosis and therapy.1–4 In turn, these applications become more demanding on the functionality and structure of nanoparticles. Ideal multifunctional nanoparticles may have the following properties: first, unique optical properties (fluorescence or localized surface plasmon resonance) or magnetism, which will be able to provide a high sensitivity and a high spectral resolution for the multiplex detection; second, a protecting shell to prevent the nanoparticles from aggregating while keeping the biocompatibility and stability under biological conditions; and third, an outer shell containing antibody, oligonucleotide, or affinity peptide to provide a specific interaction with the target species while showing a minimal toxicity.Au nanoparticles, with its special biocompatibility and unique optical properties, have been widely used in labeling, colorimetric sensing, drug delivery, biomedical treatment, etc.5–8 Up to now, direct use of bare Au nanoparticles for the above purposes may suffer from aggregation when the nanoparticles are in some detecting environments as a result of the neutralization of the surface charge. Most importantly, the competitive adsorption of solution species with the probe molecules may reduce the signal of the probe molecules or even severely interfere with the specific detection of biomolecules. Therefore, various coating methods have been developed to protect the Au nanoparticles with silica, protein, polymeretc.9–12Silica is the most widely used material for this purpose owing to its chemical inertness, easy functionalization, high stability, and optical transparency. The conventional method to form a silica coating employs a tedious three-step method: (1) surface activation for silanization with coupling silane agents, such as 3-aminopropyltrimethoxysilane (APS); (2) initial silica deposition with sodium silicate in an aqueous solution; (3) and extensive growth of the silica shell with a silicon alkoxide, such as tetraethylorthosilicate (TEOS) in an alcohol solution. Among the three steps, the second step is still not well controlled and takes a long time (from 24 h to weeks depending on the thickness) before the silica shell becomes thick enough to stabilize the particles in alcohol for the subsequent extensive growth of silica.13,14 Further improvement has been made by introducing methoxy-poly(ethylene glycol)-thiol (mPEG-SH) to the Au nanoparticle surfaces to improve their stability in the ethanol solution.15 More recently, our group have successfully shortened the time for coating the initial silica shell of 4–5 nm from sodium silicate to about one hour by elevating the reaction temperature from room temperature to 90 °C.16 However, these TEOS or sodium silicate-based SiO2 coating methods provide only silanol groups on the surface, which require further modification to introduce some specific functional groups, such as –SH, –NH2, –CHO for the subsequent binding with fluorescence molecules or biomolecules.17–19 In a typical process, the prepared SiO2 should be silanized with 3-mercaptopropyltrimethoxysilane (MPTMS), APS, or N1-[3-(trimethoxysilyl)-propyl]diethylenetriamine firstly. Then, the functionalized silica nanoparticles are conjugated with disulfide-modified oligonucleotidesvia the thiol/disulfide exchange reaction or with enzymes or antibodiesvia crosslinking between amines using glutaraldehyde.20 The multi-step procedures for synthesizing fully functionalized nanoparticles are not only time-consuming but also easy to induce the aggregation of nanoparticles.
Recently, organosilica nanoparticles made from MPTMS as the sole source prepared either through an emulsion-based route or Stöber method have found wide interests in biomolecular screening after covalently functionalized with fluorescence tags.21–25 Both the interior and exterior of organosilica nanoparticles contain exposed thiol residues, which allow the direct covalent attachment of fluorescent dyes functionalized with maleimide, isothiocyanate, succinimidyl ester, protein maleimide or disulfide-modified oligonucleotidesetc.
Due to the special optical properties of Au nanoparticles, it would be attractive if one is able to prepare Au core organosilica shell nanoparticles. It will on one hand endow the nanoparticles with the rich optical properties of Au nanoparticles and on the other hand inherit the rich functional groups of the organosilica shell. In this work, we develop a novel method to synthesize Au@organosilica nanoparticles using 3-mercaptopropyltriethoxysilane (MPS) as the sole silica source in an aqueous solution. The so-formed organosilica shell contains two different functional groups, i.e., Si–OH and –SH. The Si–OH group can covalently bind with different groups as in the normal Au@SiO2. The thiol residue provides a more convenient way to bind covalently with target molecules, such as various fluorescence molecules, proteins or single stranded DNAs (ssDNA) with isothiocyanate, succinimidyl ester or maleimide groups. If the Au core is modified with Raman reporter prior to the encapsulation of the organosilica shell, and then fluorescence molecules are introduced into the shell via covalent binding with free thiol residues, multifunctional nanoparticles with a combination of surface-enhanced Raman scattering (SERS), fluorescence and Rayleigh scattering can be obtained and used for the multimodal imaging of living cells.
Results and discussion
Preparation of Au@organosilica nanoparticles
Scheme 1A illustrates the procedure to prepare Au@organosilica nanoparticles. Au nanoparticles were firstly prepared by the seed growth method. After being modified with heterofunctional poly(ethylene glycol) (SH-PEG-COOH), the nanoparticles were further coated with an organosilica shell by hydrolysis of MPS under the basic condition. SH-PEG-COOH forms polymeric brushes on the Au nanoparticles surface to prevent the aggregation of nanoparticles for the steric reason, and therefore it plays an important role in the formation of Au@organosilica nanoparticles.12,15,26 In the absence of SH-PEG-COOH coating, Au nanoparticles will quickly form aggregates and precipitate from solution in the presence of a small amount of MPS. Fig. 1a–f show the representative TEM images of Au@organosilica nanoparticles with different shell thicknesses and the insets are the high-magnification images. The corresponding SEM images are presented in the Fig. S1, ESI†. Both the TEM and SEM results show that the Au@organosilica nanoparticles are nearly spherical and highly monodispersed. Each nanoparticle consists of a gold core with an average diameter of 55 nm and an organosilica shell with a thickness from 15 nm to 75 nm. The shell thickness could be easily adjusted by the dose of MPS. | ||
| Scheme 1 Schematic illustration of the synthetic procedure for Au@organosilica nanoparticles (A) and Au@organosilica nanoparticles with SERS and fluorescence markers together (RF-SERS nanoparticles) (B). | ||
 | ||
| Fig. 1 TEM images of Au@organosilica nanoparticles with the 55 nm Au core and different shell thicknesses: (a) 15nm, (b) 20 nm, (c) 30 nm, (d) 45 nm, (e) 60 nm, and (f) 75 nm. The insets show the nanoparticles at a high magnification. | ||
The effect of the coating on the optical response of these Au@organosilica nanoparticles is shown in Fig. 2. As can be seen in the UV-vis absorption spectra, there is an obvious red-shift of the absorption peak of Au nanoparticles after organosilica coating, which may be due to the increase of the local refractive index. This result is similar to the previous results of Au@SiO2.14
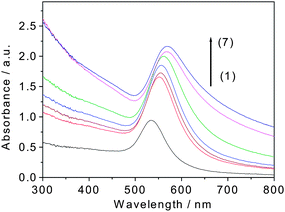 | ||
| Fig. 2 UV-vis absorption spectra of Au@organosilica nanoparticles with different shell thicknesses (1) 0 nm, (2) 15 nm, (3) 20 nm, (4) 30 nm, (5) 45 nm, (6) 60 nm, and (7) 75 nm. | ||
It is well known that Au nanoparticles form aggregates when the ionic condition of the solution is changed, resulting in a significant red shift of the plasmon absorption band to that in the separated state.27 This property was utilized to check the stability of Au nanoparticles under different ionic conditions. As expected, in the presence of organosilica shell, the stability of the nanoparticles has been significantly improved. For example, the Au@organosilica nanoparticles with a thin shell (15 nm) could be readily transferred from water into solutions of PBS, ethanol, and acetone without causing any aggregation (Fig. 3a). Whereas, the uncoated nanoparticles completely aggregate and precipitate in these solvents (Fig. 3b). The zeta potential measurements reveals that the surface charge of Au@organosilica nanoparticles was about −51.7 mV, which is much higher than that of conventional Au@SiO2nanoparticles (about −38 mV) in a control experiment. The result confirms the improved stability of the Au@organosilica nanoparticles upon organosilica shell coating. The Au@organosilica nanoparticles dispersed in deionized water can be stored for more than one year.
 | ||
| Fig. 3 UV-vis absorption spectra of Au@organosilica nanoparticles with 15 nm shell (a) and bare Au nanoparticles (b) dispersed in different solutions. | ||
In order to understand the growth mechanism of the organosilica shell, Au@organosilica nanoparticles were analyzed using the Fourier-transform infrared (FTIR), Raman and energy dispersive X-ray (EDX) spectroscopies.
FTIR spectra of Au@organosilica nanoparticles with shell thickness from 15 nm to 75 nm are shown in Fig. 4a. The band at 2563 cm−1 is from the S–H vibration, indicating that there are free SH species in the shell.28 The bands at 1125 cm−1 and 1036 cm−1 are from Si–O–Si antisymmetric stretches.29 Furthermore, the increasing intensity of S–H and Si–O–Si bands with the shell thickness reveals that the thickness of the organosilica shell can be tuned by changing the dose of MPS molecules, which agrees well with the TEM and SEM results. The number of free –SH groups was calculated according to the Ellman's reaction (the details are shown in the experiment section, ESI†), which is estimated to be about 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in each Au@organosilica nanoparticle for a shell thickness of 75 nm. The band at 295 cm−1 in the Raman spectrum (Fig. 4b) can be assigned to the Au–S vibration due to the binding of MPS and SH-PEG-COOH molecules with Au.30 Meanwhile, the band at 509 cm−1 comes from the S–S vibration, indicating the presence of the disulfide bond in the nanoparticles.31 To in situ monitor the formation of the S–S bond during the growth process of Au@organosilica nanoparticles, a roughened SERS-active Au electrode was immersed into a mixture of MPS and NH4OH solution and the SERS spectra were measured with time. No S–S band was observed before the hydrolysis, whereas the S–S band was found to increase with the hydrolysis time (Fig. S2, ESI†). EDX analysis of a single Au@organosilica nanoparticle (shell thickness around 75 nm) (Fig. 4c) shows that the atomic ratio of Si/S obtained from the peak areas is close to 1:1. All these results indicate that the –SH group plays an important role for the growth of organosilica shell. Nakamura et al. hypothesized that the MPTMS molecule, which shows an extremely similar molecular structure to MPS molecules, could make dipodal alkoxysilanes via formation of S–S bonds between the thiol residues, and the resulting dipodal alkoxysilane could produce periodic mesoporous organosilica nanoparticles.22
000 in each Au@organosilica nanoparticle for a shell thickness of 75 nm. The band at 295 cm−1 in the Raman spectrum (Fig. 4b) can be assigned to the Au–S vibration due to the binding of MPS and SH-PEG-COOH molecules with Au.30 Meanwhile, the band at 509 cm−1 comes from the S–S vibration, indicating the presence of the disulfide bond in the nanoparticles.31 To in situ monitor the formation of the S–S bond during the growth process of Au@organosilica nanoparticles, a roughened SERS-active Au electrode was immersed into a mixture of MPS and NH4OH solution and the SERS spectra were measured with time. No S–S band was observed before the hydrolysis, whereas the S–S band was found to increase with the hydrolysis time (Fig. S2, ESI†). EDX analysis of a single Au@organosilica nanoparticle (shell thickness around 75 nm) (Fig. 4c) shows that the atomic ratio of Si/S obtained from the peak areas is close to 1:1. All these results indicate that the –SH group plays an important role for the growth of organosilica shell. Nakamura et al. hypothesized that the MPTMS molecule, which shows an extremely similar molecular structure to MPS molecules, could make dipodal alkoxysilanes via formation of S–S bonds between the thiol residues, and the resulting dipodal alkoxysilane could produce periodic mesoporous organosilica nanoparticles.22
 | ||
| Fig. 4 (a) FTIR spectra of Au@organosilica nanoparticles with shell thicknesses of (1) 15 nm, (2) 20 nm, (3) 30 nm, (4) 45 nm, (5) 60 nm and (6) 75 nm. (b) Raman spectrum and (c) EDX analysis of Au@organosilica nanoparticles with a shell thickness of 75 nm. The inset in (a) is the enlarged spectra to see clearly the band at 2563 cm−1. The inset in (c) shows the TEM image of the nanoparticles used for EDX analysis and the circular region is the spot measured by EDX. The Cu signal is from the copper mesh for TEM. | ||
On the basis of the obtained results and the hypothesis of Nakamura's group, we proposed a growth mechanism of the Au@organosilica nanoparticles as follows (Fig. 5): (1) SH-PEG-COOH molecules are adsorbed on the surface of Auvia the strong Au–S interaction to form a protecting layer and stabilized the Au colloids; (2) MPS molecules fill in the vacancies at the root of the forest of the PEG molecules on the Au surface via the Au–S interaction and form the first layer of MPS; (3) the second layer of MPS molecules are conjugated with the MPS in the first layer by the condensation reaction with the –OH group under the basic condition; (4) the third layer of MPS molecules are linked with the one in the second layer by the formation of S–S bonding with the free –SH group (confirmed by the Raman band of S–S at 509 cm−1) and the three dimensional Si–O–Si network. The thiol functionalized monomer has three, rather than four reactive sites for condensation, which provides some free –SH in the whole organosilica shell (confirmed by the increasing signal of the 2563 cm−1 band in FTIR) for binding with biomarkers. Furthermore, a stronger binding affinity of the –SH groups to the Au surfaces and the Ostwald ripening effect can significantly reduce the yield of pure organosilica nanoparticlesvia homogeneous nucleation.32,33
 | ||
| Fig. 5 Proposed growth mechanism of Au@organosilica nanoparticles. | ||
Synthesis of RF-SERS Au@organosilica nanoparticles and their application for multimodal imaging
As mentioned above, ideal nanoparticles for biological applications should have multiple detection modalities (magnetism, fluorescence, elastic or inelastic light scattering), be stable under biological conditions, offer surfaces that can be easily conjugated with bio-markers, and show minimal toxicity. The effort to combine multimodal detection into one system compensates for the deficiencies of any single imaging modality and allow the fast screening of the most interesting sites on large and complicated samples, such as tissues and organs.34 For this purpose, F-SERS nanoparticle have been developed by coating a dielectric core with Raman reporter- and fluorophore-modified Ag nanoparticles and were used as a core material for biosensors based on SERS/surface-enhanced resonance Raman scattering (SERRS) and fluorescence, and significant progress has been made in this direction.35–39 For preparation of the F-SERS dots, quite tedious labeling, centrifugating and washing procedures were unavoidably used, which increase the possibility to form aggregates. The tedious procedures might be simplified by using the present method for preparing the Au@organosilica nanoparticles, which will contribute to the further development and extension of F-SERS. In addition, the present method not only offers a metal core with a very strong Rayleigh scattering signal, well exceeding that of SERS and fluorescence, but also significantly improves the photostability of the fluorophores by incorporating them inside the shell. Using these nanoparticles, we can use strong Rayleigh scattering for fast screening at the video rate, confocal fluorescence for profiling interesting species, and SERS for multiplex analysis. The nanoparticles with these modalities can be named as RF-SERS dots.Scheme 1B shows the route for preparing RF-SERS Au@organosilica nanoparticles. Au cores were modified with Raman probe molecules, malachite green isothiocyanate (MGITC), before the encapsulation of the organosilica shell. The concentration of the Raman reporter molecules were controlled at ca. 10−8 M, which is less than that needed for formation of a monolayer on Au nanoparticles. The labelled nanoparticles were then coated with organosilica shells following the above mentioned procedure. The organosilica shells were further conjugated with fluorescence molecules such as fluorescein isothiocyanate (FITC) by a simple mixing. Multifunctional nanoparticles (RF-SERS NPs), showing SERS, fluorescence, and Rayleigh scattering, were then obtained and used for the multimodal imaging of living cells.
We chose the MGITC and FITC as Raman probe and fluorescence molecule respectively for the following three reasons. First, MGITC has an absorption ca. 629 nm, close to the laser line (632.8 nm) we used,40 which can produce SERRS with a signal that is 2–3 orders higher than that of SERS alone. Secondly, MGITC with an anchoring isothiocyanate (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS) group provides an “affinity tag” for binding to Au surfaces, yielding a S–Au bond that is stable against the coupling agent and organosilica deposition.41 Finally, the emission of FITC is around 520 nm and it has only minor interference with the SERS signal excited with 632.8 nm laser. From the SEM and TEM images of MGITC labelled Au@organosilica nanoparticles, the presence of sub-monolayer of MGITC molecules has a minor influence on the monodispersity of Au@organosilica nanoparticles at a shell thickness up to 30 nm. At a thickness of 45 nm and above, some gold aggregates can be identified (Fig. S3, ESI†). Therefore, in the following studies, the MGITC labelled Au@organosilica nanoparticles with shell thickness of 30 nm were used for multimodal image to avoid the aggregation. If a thicker shell is desired, a stepwise way to add MPS can successfully reduce the aggregation for MGITC labelled Au@organosilica nanoparticles but it will reduce the simplicity of the present method.
CS) group provides an “affinity tag” for binding to Au surfaces, yielding a S–Au bond that is stable against the coupling agent and organosilica deposition.41 Finally, the emission of FITC is around 520 nm and it has only minor interference with the SERS signal excited with 632.8 nm laser. From the SEM and TEM images of MGITC labelled Au@organosilica nanoparticles, the presence of sub-monolayer of MGITC molecules has a minor influence on the monodispersity of Au@organosilica nanoparticles at a shell thickness up to 30 nm. At a thickness of 45 nm and above, some gold aggregates can be identified (Fig. S3, ESI†). Therefore, in the following studies, the MGITC labelled Au@organosilica nanoparticles with shell thickness of 30 nm were used for multimodal image to avoid the aggregation. If a thicker shell is desired, a stepwise way to add MPS can successfully reduce the aggregation for MGITC labelled Au@organosilica nanoparticles but it will reduce the simplicity of the present method.
Fig. 6a shows the SERS spectra of MGITC labelled Au@organosilica nanoparticles with different shell thicknesses in sol. The characteristic bands of MGITC (1171, 1217, 1294, 1363, 1387 and 1613 cm−1) can be clearly identified, which demonstrated that MGITC had not been replaced during the coating of the organosilica shell. The intensity does not vary obviously when the shell is thinner than 45 nm. However, at a larger thickness, the intensity of the SERS signal slightly decreases with the increase of the thickness, which can be attributed to a lower transmission of light at a thicker organosilica shell. To confirm that the FITC molecules have been bound to the MGITC labelled Au@organosilica nanoparticles, fluorescence emission spectra (excitation at 488 nm) of FITC-MGITC Au@organosilica nanoparticles with different shell thicknesses in colloidal solutions were studied (Fig. 6b). The emission spectra of free FITC, MGITC solutions (dashed curve in Fig. 6b) and the colloid solutions of bare Au@organosilica nanoparticles with different organosilica shell thicknesses (Fig. 6c) were also given for comparison. FITC-MGITC Au@organosilica nanoparticles were centrifuged and washed with deionized water several times until there was no detectable fluorescence from the supernatant. It can be seen from the fluorescence spectra shown in Fig. 6b that the fluorescence intensity increases with the increasing shell thickness, indicating more FITC molecules are in the shell. No emission signal was observed under the excitation of 488 nm in the free MGITC solution (Fig. 6b). The emission spectra (Fig. 6c) of bare Au@organosilica nanoparticles are contributed by the Au core and experience minor change with the increase of the organosilica shell thickness. The results show that FITC molecules can easily be incorporated into the organosilica shell via covalent bonding between free thiol group and isothiocyanate group, which significantly reduces the release of FITC dye from Au@organosilica nanoparticles (see Fig. S4, ESI† for release curve) literatures.21,42 The obtained RF-SERS Au@organosilica nanoparticles can be further used for the cell imaging.
 | ||
| Fig. 6 (a) SERS of MGITC labelled Au@organosilica nanoparticles with different shell thicknesses as indicated in the figure. Fluorescence spectra of free FITC, MGITC solutions, and FITC-MGITC Au@organosilica nanoparticles with different organosilica shell thicknesses in colloidal solutions (b) and Au@organosilica nanoparticles (c) as a comparison (excitation at 488 nm, emission was detected at 500–600 nm). | ||
The cytotoxicity behavior will be the major concern before the FITC-MGITC Au@organosilica nanoparticles can be used for living cell imaging. Therefore, we investigate the effect of the dose and size of RF-SERS Au@organosilica nanoparticles on their cytotoxicity to choose the optimal condition for the multimodal imaging of living cells. We used the established MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay to investigate the cytotoxicity of FITC-MGITC Au@organosilica nanoparticles on HeLa cells. FITC-MGITC Au@organosilica nanoparticles show a dose-dependent toxicity after incubated with cells for 24 h (Fig. 7). No obvious cytotoxicity was observed at a lower dose, and all the nanoparticles inevitably show some toxicity at a very high dose. More interestingly, the cytotoxicity of monodispersed FITC-MGITC Au@organosilica nanoparticles with a same morphology strongly depends on the particle size. Smaller particles show a significantly higher toxicity than the bigger ones when the dose is expressed in mass concentration. The result is similar to the previous work about the cytotoxicity of SiO2.43 Considering that the upper size limit for an efficient uptake through the cell membrane appears to be around 100 nm 44 and the cytotoxicity of Au@organosilica nanoparticles will increase at smaller sizes, we chose FITC-MGITC Au@organosilica nanoparticles with 30 nm shell thickness (total size is about 115 nm) for the multimodal application. For this purpose, nanoparticles at a final concentration of 30 μg mL−1 (show almost no cytotoxicity at this concentration confirmed by MTT assay) in the media without bovine serum albumin were incubated with HeLa cells at 37 °C for 12 h. The cells were then stained with Hoechst 33258 to visualize the cell nucleus (blue). Subsequently, they were analyzed by dark field microscopy, confocal laser scanning microscopy (CLSM), and SERS (Fig. 8).
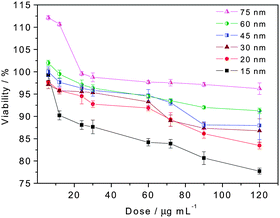 | ||
| Fig. 7 Viability of HeLa cells after 24 h exposure to the increasing dose of FITC-MGITC Au@organosilica nanoparticles with different shell thicknesses (15, 20, 30, 45, 60, and 75 nm). | ||
 | ||
| Fig. 8 Confocal microscopy image (a) and dark field microscopic image (b) showing the distribution of FITC-MGITC Au@organosilica nanoparticles with 30 nm shell thickness in cell. Nanoparticles appear green, nuclei stained with Hoechst 33258 appear blue. (c) SERS spectra of the FITC-MGITC Au@organosilica nanoparticles from different regions of the cell marked in (b). | ||
The strong Rayleigh scattering of the Au core allows the direct imaging of FITC-MGITC Au@organosilica nanoparticles at a video rate using a normal white light illumination under the dark-field configuration. As the Rayleigh scattering process can produce signals much stronger than the fluorescence process,45 the images are free of interference of the autofluorescence of living cells. However, due to the wide field imaging configuration used in the dark field microscopy, it lacks the z-direction resolution inherent with the confocal microscopy. Therefore, it is not straightforward to distinguish whether the nanoparticles are just adhered on the cell membrane through the nonspecific interactions or inside the cell through endocytosis. Nevertheless, the nanoparticles with Au cores can be very efficiently used for the fast screening purpose to easily locate the position and determine the quantity of nanoparticles in the whole sample without the interference of the autofluorescence of the biological sample by using the dark-field imaging. Therefore, it is beneficial for the more complicated sample such as tissue and in vivo imaging.
CLSM, on the other hand, can provide very high spatial resolution in the vertical direction. After fast screening with Rayleigh scattering, we then used CLSM to more accurately determine the subcelluar distribution of the nanoparticles in the cell. For this purpose, merged CLSM images of HeLa cell were taken (Fig. 8a). Nucleus and the nanoparticles appear as blue and green in the image. As can be seen in the CLSM images, the FITC-MGITC Au@organosilica nanoparticles can be internalized into HeLa cells, and the ingested nanoparticles are distributed mostly in the cytoplasm region and no nanoparticles are found in the nuclei region. Though in the absence of any receptor moieties on the cell membrane surface, FITC-MGITC Au@organosilica nanoparticles can still be ingested by HeLa cells, indicating that a nonreceptor-mediated endocytosis pathway exists in the HeLa cells, which agrees with the literature.46SERS is advantageous and complementary to fluorescence spectroscopy for multiplex detection for the following obvious reasons:9,37–39,47–52 the full width at half maximum (FWHM) of SERS bands (∼1 nm),53,54 is much narrower than that of most fluorophores (50–100 nm)53,55 or quantum dots (25–40 nm),56 minimizing the possible overlapping of different label molecules in a given spectral range; complicated statistics and deconvolution, required to resolve multiple overlapped fluorescence spectra can be avoided by using SERS; SERS probes can be chosen from a wide range of labeling candidates including both fluorophores and nonfluorophores;48,57 photobleaching and blinking behavior suffered by fluorescent labels can be avoided; and SERS can be generated using far-infrared or near-infrared lasers with energies much less than that necessary to excite the inherent background fluorescence in biological samples. We then used SERS to follow the Raman signal of the sample. Fig. 8c shows the SERS spectra MGITC obtained on different spots of the dark field image (Fig. 8b) and no obvious interference from the labelled fluorescent FITC molecules. The SERS signals clearly show a distribution of the nanoparticles in the cell and can be continuously tracked without the bother of photo-bleaching or photodegradation.
To further demonstrate the multiplex capability of the Au@organosilica nanoparticles, MGITC and X-rhodamine-5-(and-6)-isothiocyanate (XRITC) are chosen as probe molecules. The 1:1 mixture of the as-prepared FITC-MGITC Au@organosilica nanoparticles and FITC-XRITC Au@organosilica nanoparticles were added into the culture media and incubated with the HeLa cell for 12 h. After fixing, washing, and nucleus staining, the sample was studied by CLSM and SERS. Fig. 9a shows the SERS spectra of FITC-MGITC Au@organosilica nanoparticles, FITC-XRITC Au@organosilica nanoparticles, and their 1:1 mixture from top to bottom. The signals of MGITC and XRITC can be easily distinguished in the mixture. Fig. 9b shows the emerged fluorescence image of nanoparticles using FITC as the probe and the stained nucleus using Hoechst 33258 as the probe. Similar to previous results, the nanoparticles can be ingested into the cell plasma but not the nucleus. We then focused on one cell with the clear feature of nanoparticles (see Fig. 9c, appears as yellow to orange spots) for multiplex two dimensional imaging using the 632.8 nm excitation. In each pixel, a spectrum with a spectral range from about 1200 to 1800 cm−1 was obtained. Then, we took the two bands at 1613 and 1502 cm−1, characteristic of MGITC and XRITC respectively to construct Raman images, and the result is shown in Fig. 9d and 9e. To faithfully reflect the distribution of the nanoparticles and the scanning step, we present the images by pixels rather than the interpolated and smoothed image treated by the software. The emerged image of the two figures is shown in Fig. 9f. It can be seen from the three images that the two types of nanoparticles distribute differently in the cell. When we picked out the SERS spectra (Fig. 9g) from some characteristic points, we found a distribution of nanoparticles in the cell. For example, at points 1 and 3, we could only detect one type of nanoparticle. The above result indicates that RF-SERS Au@organosilica nanoparticles can be used for multiplex detection and imaging.
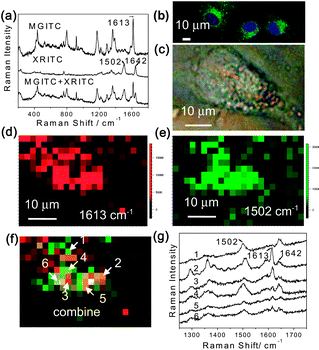 | ||
| Fig. 9 Au@organosilica nanoparticles for multiplex detection. (a) SERS spectra of FITC-MGITC Au@organosilica nanoparticles, XRITC-MGITC Au@organosilica nanoparticles, and their 1:1 mixture. (b) Confocal microscopic fluorescence images showing the distribution of FITC-MGITC Au@organosilica nanoparticles and FITC-XRITC Au@organosilica nanoparticles in the cell. (c) Bright field microscopic image of a HeLa cell. SERS images produced by using the baseline corrected intensity of the 1613 cm−1 Raman band of MGITC (d) and the 1502 cm−1 Raman band of XRITC (e) respectively. (f) The merged figure of (d) and (e) to illustrate the distribution of two types of labelled multifunctional nanoparticles in the cell. (g) Typical SERS spectra obtained at different locations of (f). | ||
Conclusions
In summary, we have developed a method to synthesize discrete and monodispersed Au@organosilica nanoparticles by using a simple procedure from a sole silica source-MPS in an aqueous solution. This method has been successfully used to synthesize multifunctional nanoparticles with Raman reporters adsorbed directly on the Au surface and the fluorescence molecules embedded within the organosilica shell. The formed RF-SERS multifunctional nanoparticles have been used for multimodal imaging of living cells with the unique optical properties of Rayleigh scattering of the Au core, fluorescence of the dye molecule, and SERRS of the resonance Raman molecules. By choosing resonance Raman molecules with well separated bands, multiplex imaging using the SERS signal has been successfully demonstrated revealing the promising future of such a kind of multifunctional nanoparticles. Further study using different optical properties of the nanoparticles may reveal important information of the dynamic interaction between the nanoparticles and the living cell with a proper design of the surface functional molecules, such as receptor. This simple synthetic strategy for obtaining organosilica-encapsulated nanoparticles in the aqueous solution can be further extended to other nanoparticles, such as magnetic nanoparticles and luminescent quantum dots. With further development of the microscopic and spectroscopic instruments, the integration of SERS, CLSM, and darkfield microscope into one system can be easily realized. Then, the potential of such a kind of multifunctional nanoparticles can be fully exerted to provide abundant information of systems of biological importance.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program nos. 2007CB935603, 2009CB930703, and 2007DFC40440) and the Natural Science Foundation of China (Nos. 20620130427, 20825313, and 20827003, 21021120456 and 21021002).Notes and references
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS.
- A. Gole, N. Agarwal, P. Nagaria, M. D. Wyatt and C. J. Murphy, Chem. Commun., 2008, 6140–6142 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- M. D. Porter, R. J. Lipert, L. M. Siperko, G. Wang and R. Narayanana, Chem. Soc. Rev., 2008, 37, 1001–1011 RSC.
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- A. M. Schwartzberg, T. Y. Oshiro, J. Z. Zhang, T. Huser and C. E. Talley, Anal. Chem., 2006, 78, 4732–4736 CrossRef CAS.
- W. E. Doering, M. E. Piotti, M. J. Natan and R. G. Freeman, Adv. Mater., 2007, 19, 3100–3108 CrossRef CAS.
- B. Kustner, M. Gellner, M. Schutz, F. Schoppler, A. Marx, P. Strobel, P. Adam, C. Schmuck and S. Schlucker, Angew. Chem., Int. Ed., 2009, 48, 1950–1953 CrossRef.
- X. Su, J. Zhang, L. Sun, T. W. Koo, S. Chan, N. Sundararajan, M. Yamakawa and A. A. Berlin, Nano Lett., 2005, 5, 49–54 CrossRef CAS.
- X. M. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. M. Nie, Nat. Biotechnol., 2007, 26, 83–90.
- L. M. Liz-Marzan and P. Mulvaney, J. Colloid Interface Sci., 1995, 176, 459–466 CrossRef CAS.
- L. M. Liz-Marzan, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef CAS.
- C. Fernandez-Lopez, C. Mateo-Mateo, R. A. Alvarez-Puebla, J. Perez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Langmuir, 2009, 25, 13894–13899 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 464, pp. 392–395 Search PubMed.
- H. B. Shen, X. S. Yang, M. Y. Gao, N. Q. Jia, F. Wang and N. Zhao, Chem. Mater., 2007, 19, 3090–3092 Search PubMed.
- S. H. Liu and M. Y. Han, Adv. Funct. Mater., 2005, 15, 961–967 CrossRef CAS.
- J. L. Gong, Y. Liang, Y. Huang, J. W. Chen, J. H. Jiang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2007, 22, 1501–1507 CrossRef CAS.
- D. Knopp, D. P. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14–30 CrossRef CAS.
- A. P. R. Johnston, B. J. Battersby, G. A. Lawrie and M. Trau, Chem. Commun., 2005, 848–850 RSC.
- M. Nakamura and K. Ishimura, J. Phys. Chem. C, 2007, 111, 18892–18898 CrossRef CAS.
- C. R. Miller, R. Vogel, P. P. T. Surawski, K. S. Jack, S. R. Corrie and M. Trau, Langmuir, 2005, 21, 9733–9740 CrossRef CAS.
- S. R. Corrie, G. A. Lawrie and M. Trau, Langmuir, 2006, 22, 2731–2737 CrossRef CAS.
- M. Nakamura and K. Ishimura, Langmuir, 2008, 24, 5099–5108 CrossRef CAS.
- A. G. Kanaras, F. S. Kamounah, K. Schaumburg, C. J. Kiely and M. Brust, Chem. Commun., 2002, 2294–2295 RSC.
- G. R. Souza, D. R. Christianson, F. I. Staquicini, M. G. Ozawa, E. Y. Snyder, R. L. Sidman, J. H. Miller, W. Arap and R. Pasqualini, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1215–1220 CrossRef CAS.
- G. Colotti, E. Chiancone and A. Boffi, Biochemistry, 1999, 38, 10079–10083 Search PubMed.
- J. M. Antonucci, S. H. Dickens, B. O. Fowler, H. H. K. Xu and W. G. McDonough, J. Res. Natl. Inst. Stand. Technol., 2005, 110, 541–558 Search PubMed.
- X. Gao, Y. Zhang and M. J. Weaver, Langmuir, 1992, 8, 668–672 CrossRef CAS.
- Y. Ozaki, A. Mizuno, K. Itoh and K. Iriyama, J. Biol. Chem., 1987, 262, 15545–15551 Search PubMed.
- N. G. Liu, B. S. Prall and V. I. Klimov, J. Am. Chem. Soc., 2006, 128, 15362–15363 CrossRef CAS.
- W. Ostwald, Z. Phys. Chem, 1900, 34, 495–499 Search PubMed.
- M. V. Yigit, L. Y. Zhu, M. A. Ifediba, Y. Zhang, K. Carr, A. Moore and Z. Medarova, ACS Nano, 2011, 5, 1056–1066 Search PubMed.
- K. Kim, H. B. Lee, Y. M. Lee and K. S. Shin, Biosens. Bioelectron., 2009, 24, 1864–1869 CrossRef CAS.
- K. Kim, Y. M. Lee, H. B. Lee and K. S. Shin, Biosens. Bioelectron., 2009, 24, 3615–3621 CrossRef CAS.
- B. H. Jun, J. H. Kim, H. Park, J. S. Kim, K. N. Yu, S. M. Lee, H. Choi, S. Y. Kwak, Y. K. Kim, D. H. Jeong, M. H. Cho and Y. S. Lee, J. Comb. Chem., 2007, 9, 237–244 CrossRef CAS.
- K. N. Yu, S. M. Lee, J. Y. Han, H. Park, M. A. Woo, M. S. Noh, S. K. Hwang, J. T. Kwon, H. Jin, Y. K. Kim, P. J. Hergenrother, D. H. Jeong, Y. S. Lee and M. H. Cho, Bioconjugate Chem., 2007, 18, 1155–1162 CrossRef CAS.
- M. A. Woo, S. M. Lee, G. Kim, J. Baek, M. S. Noh, J. E. Kim, S. J. Park, A. Minai-Tehrani, S. C. Park, Y. T. Seo, Y. K. Kim, Y. S. Lee, D. H. Jeong and M. H. Cho, Anal. Chem., 2009, 81, 1008–1015 CrossRef CAS.
- Z. Q. Tian, B. Ren, J. F. Li and Z. L. Yang, Chem. Commun., 2007, 3514–3534 RSC.
- W. E. Doering and S. M. Nie, Anal. Chem., 2003, 75, 6171–6176 CrossRef CAS.
- Y. H. Chan, C. Wu, F. Ye, Y. Jin, P. B. Smith and D. T. Chiu, Anal. Chem., 2011, 83, 1448–1455 Search PubMed.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846–853 CrossRef CAS.
- V. Sokolova and M. Epple, Angew. Chem., Int. Ed., 2008, 47, 1382–1395 CrossRef CAS.
- S. Schultz, D. R. Smith, J. J. Mock and D. A. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 996–1001 CrossRef CAS.
- I. P. Chang, K. C. Hwang and C. S. Chiang, J. Am. Chem. Soc., 2008, 130, 15476–15481 CrossRef CAS.
- L. Sun, K. B. Sung, C. Dentinger, B. Lutz, L. Nguyen, J. W. Zhang, H. Y. Qin, M. Yamakawa, M. Q. Cao, Y. Lu, A. J. Chmura, J. Zhu, X. Su, A. A. Berlin, S. Chan and B. Knudsen, Nano Lett., 2007, 7, 351–356 CrossRef CAS.
- L. Sun, C. X. Yu and J. Irudayaraj, Anal. Chem., 2008, 80, 3342–3349 CrossRef CAS.
- A. MacAskill, D. Crawford, D. Graham and K. Faulds, Anal. Chem., 2009, 81, 8134–8140 CrossRef CAS.
- G. F. Wang, H. Y. Park and R. J. Lipert, Anal. Chem., 2009, 81, 9643–9650 CrossRef CAS.
- C. I. Brady, N. H. Mack, L. O. Brown and S. K. Doorn, Anal. Chem., 2009, 81, 7181–7188 Search PubMed.
- H. N. Wang and T. Vo-Dinh, Nanotechnology, 2009, 20, 6.
- J. Ni, R. J. Lipert, G. B. Dawson and M. D. Porter, Anal. Chem., 1999, 71, 4903–4908 CrossRef CAS.
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef.
- N. R. Isola, D. L. Stokes and T. Vo-Dinh, Anal. Chem., 1998, 70, 1352–1356 CrossRef CAS.
- C. Y. Zhang and L. W. Johnson, J. Am. Chem. Soc., 2006, 128, 5324–5325 CrossRef CAS.
- F. T. Docherty, M. Clark, G. McNay, D. Graham and W. E. Smith, Faraday Discuss., 2004, 126, 281–288 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00242b |
| This journal is © The Royal Society of Chemistry 2011 |