An improved catalyst architecture for rhodium(III) catalyzed C–H activation and its application to pyridone synthesis†
Todd K.
Hyster
and
Tomislav
Rovis
*
Department of Chemistry, Fort Collins, CO 80523, USA. E-mail: rovis@lamar.colostate.edu; Tel: +970-491-7208
First published on 16th May 2011
Abstract
We have developed a method for preparing pyridones from the coupling reaction of acrylamides and alkynes with either stoichometric Cu(OAc)2 or catalyticCu(OAc)2 and air as oxidants. In the course of these studies, it was found that a larger ligand, 1,3-di-tert-butylcyclopentadienyl (termed Cpt) results in higher degrees of regioselectivity in the alkyneinsertion event. The transformation tolerates a broad variety of alkynes and acrylamides. Furthermore, Cpt and Cp* demonstrate similar catalytic activity. This similarity allows for mechanistic studies to be undertaken which suggest a difference in mechanism between this reaction and the previously studied benzamide system.
Ligand development is a key element in the growth of organometallic catalysis. While directed C–H activation has experienced an explosion of interest in the last decade, few groups have tackled ligand development to increase reactivity and selectivity. Exceptions include Yu and co-workers who have developed a variety of amino acid based ligands to increase the reactivity,1 regioselectivity, and enantioselectivity2 of a transformation. Vedernikov and co-workers found 2,6-pyridinedicarboxylic acids facilitate the aerobic oxidation of 8-methylquinoline.3 Sanford and Arnold have found carbene ligands can stabilize Pd(IV) intermediates in directed halogenations.4Rh(III) C–H activation, which has been used for the synthesis of a variety of heterocycles,5 has seen very little ligand development. A prominent exception is the introduction of polyphenylated cyclopentadienyl ligand on Rh(III) by Satoh and Miura.6
 | (1) |
 | (2) |
Previously, our group7 and others8 employed Rh(III) C–H activation in the preparation of isoquinolones from benzamides and alkynes, eq (1). The reaction was proposed to proceed via aryl C–H activation by Rh(III) followed by alkyneinsertion. As we looked to expand this reaction to the more elusive alkene sp2 C–H activation with acrylamides in order to access pyridones, a ubiquitous scaffold in biologically relevant molecules,9 we found low regioselectivities for the insertion of unsymmetrical alkynes (eq (2)). Herein we disclose our development of a new ligand for Rh(III)-catalyzed C–H activation, which leads to significantly improved regioselectivity of alkyneinsertionen route to pyridone products. We further describe a series of mechanistic studies, which shed considerable light on the mechanism of this transformation.
Building on our own work, we began our investigations in the oxidative coupling of acrylamide and alkynes.10 Employing conditions used in our isoquinolone synthesis affords the desired pyridone3a in 30% yield and 7.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity (entry 1, Table 1). A brief solvent screen revealed dichloroethane to be an ideal solvent with 82% yield and identical regioselectivity (entry 2, Table 1). The desire for milder conditions directed us to [RhCp*(MeCN)3](SbF6)2, a catalyst used with success by Fagnou and co-workers.11 With this catalyst the reaction can be run at temperatures ranging from 60 °C to 110 °C with good yields but a moderate decrease in regioselectivity to 6:1. In our previous work with benzamides, the insertion of unsymmetrical alkynes universally proceeds with regioselectivities >10:1; however, with acrylamides, the selectivities are significantly lower. We speculated that a modification of the ligand might increase the selectivity of alkyneinsertion. To that end, we synthesized a Rh(III) precatalyst bearing a 1,3-di-tert-butyl cyclopentadienyl group (Cpt), similar to a species previously described by Jin.12 Gratifyingly, the use of this catalyst results in an improved regioselectivity with no loss in yield (contrast Cp* and Cpt columns, Table 1). Stimulated by this result, we conducted a screen of various acrylamide and alkyne-coupling partners under these conditions.
:1 regioselectivity (entry 1, Table 1). A brief solvent screen revealed dichloroethane to be an ideal solvent with 82% yield and identical regioselectivity (entry 2, Table 1). The desire for milder conditions directed us to [RhCp*(MeCN)3](SbF6)2, a catalyst used with success by Fagnou and co-workers.11 With this catalyst the reaction can be run at temperatures ranging from 60 °C to 110 °C with good yields but a moderate decrease in regioselectivity to 6:1. In our previous work with benzamides, the insertion of unsymmetrical alkynes universally proceeds with regioselectivities >10:1; however, with acrylamides, the selectivities are significantly lower. We speculated that a modification of the ligand might increase the selectivity of alkyneinsertion. To that end, we synthesized a Rh(III) precatalyst bearing a 1,3-di-tert-butyl cyclopentadienyl group (Cpt), similar to a species previously described by Jin.12 Gratifyingly, the use of this catalyst results in an improved regioselectivity with no loss in yield (contrast Cp* and Cpt columns, Table 1). Stimulated by this result, we conducted a screen of various acrylamide and alkyne-coupling partners under these conditions.

We were pleased to find that extensive substitution on the acrylamide is tolerated for pyridone formation. AlkylN-substituted acrylamides are tolerated with high yield and high regioselectivity (Table 2, entries 1, 2, 8). The syntheses of amino acid protected pyridones are important for medicinal targets.13 The cinnamamide derived from alanine methyl ester is tolerated in the reaction, and delivers the pyridone in high yield and regioselectivity (Table 2, entry 3). Alkyl substitution on the alkene (Table 2, entries 4–7) results in high yield but lackluster regioselectivity; however, this can be improved when Cpt is used. The use of an acrylamide containing an enamide results in formation of pyridone exclusively with good levels of selectivity and no pyrrole formation14 (Table 2, entry 8). Finally, a variety of aryl substitution on the double bond is accepted in all cases resulting in high selectivity using Cp* (Table 2, entries 10–12).

An investigation of the alkyne component reveals a variety of substitution patterns are tolerated. Propiolates are tolerated when excess alkyne is used resulting in pyridone formation in good yield and regioselectivity (Table 2, entry 13). Unsymmetrical alkynes with alkyl and aryl substitution engage with good yields and selectivity that can be enhanced using Cpt (Table 2, entries 14 and 17). Protected propargyl amines and alcohols are permitted, while their unprotected counterparts result in no product formation (Table 2, entries 15 and 16). Additionally, di-alkyl alkynes with sterically different groups result in product formation in high regioselectivity whereas di-alkyl alkynes with small differences in the sterics of two groups result in much lower selectivity (Table 2, entries 19 and 20). Enynes were employed in this reaction with high levels of selectivity, which one could imagine would afford access to the products of di-alkyl alkynes with the opposite regioselectivity after olefinreduction (Table 2, entry 18).15 In light of the propensity of terminal alkynes to dimerize, we imagined that silyl protected alkynes would render an acceptable solution. TMS protected phenylacetylene results in dimerization with a small degree of product formation. The more robust TES protected alkynes function better with moderate yield and good levels of selectivity (Table 2, entry 21). It is important to note that Cpt does not have a beneficial effect on selectivity in the cases of TES-protected alkynes.16
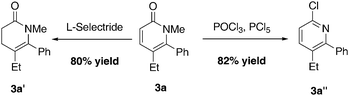
While pyridones are a common motif, it would be advantageous to derivatize these products to access different nitrogen containing heterocycles (Scheme 1). Chloropyridine3a′′, a potentially useful cross-coupling partner for further elaboration, can be accessed in 82% yield when pyridone3a is treated with POCl3/PCl5. The dihydro pyridone scaffold can be prepared in 80% yield when pyridone3a is treated with L-Selectride.
 | ||
| Scheme 1 Derivatization | ||
Inspection of our conditions led us to realize that this process would be less acceptable for large scale reactions (Table 3). Specifically, while we felt reasonably confident that Rhcatalyst loading could be decreased on scale, the use of 2 equivalents of Cu(OAc)2 as a stoichiometric oxidant delivers unneeded excess copper waste. In an effort to resolve these issues, we looked to decrease the catalyst and oxidant loading. We were pleased to find that when the oxidant loading is decreased to 20 mol% and the reaction is run open to air, pyridone formation is observed in >90% yield; however, subsequent decrease in the catalyst loading to 1 mol % forms pyridone in only 15% yield. Portionwise addition of alkyne over 30 h improves the yield to 50%. This suggests an inhibitory affect by the alkyne.17 Furthermore, an increase in the concentration of the reaction delivers higher conversions (entry 3 vs. 4, Table 3) and a slightly higher Rhcatalyst loading provides optimal results (entry 6, 6 mmol scale). Preliminary experiments suggest copper is not needed at all if the reaction is conducted under oxygen in the presence of NaOAc as base (entry 7, Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Scale (mmol) | Catalyst (mol %) | Conc. (M) | Cu(OAc)2·H2O (mol %) | GC-MS yield (%) |
| a Portionwise addition of alkyne; see ESI.† b Isolated yield. c Reaction conducted under O2 in presence of NaOAc (4 equiv). | |||||
| 1 | 0.22 | 5 | 0.3 | 20 | 86 |
| 2 | 0.22 | 1 | 0.3 | 20 | 15 |
| 3a | 0.22 | 1 | 0.3 | 20 | 37 |
| 4a | 0.22 | 2.5 | 0.15 | 20 | 35 |
| 5a | 0.44 | 1 | 0.6 | 20 | 45 |
| 6a | 6.22 | 2 | 0.6 | 20 | 69b |
| 7c | 0.22 | 5 | 0.3 | 0 | 40 |

The variation in regioselectivity as a function of substrate was perhaps most puzzling (witness 3a, 3c, 3h, 3i in Table 2 compared to the corresponding reaction with N-methyl benzamide which gives >20:1 regioselectivity). The simplest explanation for this dichotomy is a change in mechanism. Pointed experiments were conducted to try to shed light on this issue.18
 | (3) |
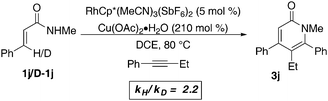
In an effort to probe whether C–H activation is the turnover-limiting step, we set out to determine the kinetic isotope effect. Using initial rates kinetics with proteo/deutero N-methylcinnamide a KIE value of 2.2 is observed. While this value fits with a primary kinetic isotope effect, and is within the range of KIE values found in palladium and rhodium catalyzed C–H activations,19,20 we felt further evidence was required before proposing C–H activation as the turnover limiting step. If C–H insertion is the turnover-limiting step, then an increase in the acidity of the cleavable proton should have a positive effect on the rate of the reaction. Consequently we employed a competition experiment between N-methylcinnamamide and the N-methyl-pentafluorophenyl-cinnamamide derivative 1n, using a 1:1:1 stoichiometry between the two amides and the alkyne. The reaction proved counter-intuitive delivering 3j as the only observed pyridone product.
 | (4) |
This surprising finding prompted us to conduct a Hammett study. A variety of substituted cinnamides were prepared and their incorporation in the reaction was compared to the parent cinnamide1j. The Hammett plot reveals a potential break, with more electron-rich and more electron-deficient systems both undergoing reaction slower than electron-neutral substrates.21 To provide further support for this hypothesis, we conducted a kinetic isotope effect study with the two extremes of reactivity.
 | (5) |
Clearly, the kinetic isotope effect data and the competition experiment described above do not uniformly support the CMD mechanism.21 We propose the following mechanism (Scheme 2). The pre-catalyst presumably loses an acetonitrile ligand to form an acetate-ligated species. Coordination of an equivalent of acrylamide forms a cationic species A. This intermediate can then undergo acetate assisted C–H activation to render intermediate B.22 The 5-membered rhodacycle B can experience a regioselective insertion of one equivalent of alkyne, with the improved regioselectivity associated with Cpt a consequence of the extreme steric differentiation between a tert-butyl group and a hydrogen. Based of the work of Jones and co-workers,23 it is likely that the C–Rh bond migrates to access the 7-membered rhodacycle C. At this point reductive elimination can occur to generate the desired pyridone and a rhodium(I) species. This species can undergo two single electron oxidationsviaCu(OAc)2 to render the catalytically active rhodium species. The Hammett plot and isotope effect study suggest that for electron-rich substrates, C–H activation is turnover limiting while in the case of the more acidic substrates, a subsequent step is turnover limiting, either alkyneinsertion or reductive elimination.
 | ||
| Scheme 2 Proposed mechanism | ||
Conclusions
We have described a new ligand for rhodium(III) C–H activation. This ligand has the benefit of increasing the regioselectivity of the alkyneinsertion event, a problem common to Rh(III) C–H activation. Additionally, we have found that the mechanism of the transformation is not uniform with its benzamide counterpart, and provides evidence for a distinct change in mechanism as a function of substrate electronics.Acknowledgements
We thank NIGMS for generous support of this research (GM80442). T.R. thanks Amgen and Roche for unrestricted support. We thank Johnson Matthey for a generous loan of Rh salts.Notes and references
-
(a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS
; (b) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS
-
(a) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS
- J. Zhang, E. Khaskin, N. P. Anderson and A. N. Vedernikov, Chem. Commun., 2008, 3625 RSC
- P. L. Arnold, M. S. Sanford and S. M. Pearson, J. Am. Chem. Soc., 2009, 131, 13912 CrossRef CAS
- For a review of rhodium(III) C–H activation, see: T. Satoh and M. Miura, Chem.–Eur. J., 2010, 18, 11212 Search PubMed
-
(a) M. Shimizu, H. Tsurugi, T. Satoh and M. Miura, Chem.–Asian J., 2008, 3, 881 CrossRef CAS
- T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS
-
(a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS
-
(a) L. A. Mitscher, Chem. Rev., 2005, 105, 559 CrossRef CAS
- During the preparation and submission of this manuscript Li and co-worker demonstrated rhodium(III) catalyzed pyridone formation with α-substituted acrylamides; see: Y. Su, M. Zhao, G. Song and X. Li, Org. Lett., 2010, 12, 5462 Search PubMed
-
(a) D. J. Schipper, M. Hutchinson and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6910 CrossRef CAS
- Q.-A. Kong and G.-X. Jin, Yingyong Huaxue, 2001, 18, 322 Search PubMed
- Y.-Q. Fang, M. M. Bio, K. B. Hansen, M. S. Potter and A. Clausen, J. Am. Chem. Soc., 2010, 132, 15525 Search PubMed
-
(a) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS
- During the preparation of this manuscript Huestis/Fagnou reported the use of enynes in the synthesis of indoles and pyrrole resulting in apparent reversed regioselectivity upon hydrogenation; see ref. 11c.
- Stuart/Fagnou report the use of TMS protected alkynes; see ref. 11d.
- This was observed by Stuart/Fagnou; see ref. 11d.
- To probe the necessity of a cis-β-proton, the N-methyl amide derived from angelic acid was prepared. When this amide is submitted to the reaction conditions at extended reaction times and elevated temperatures, none of the desired pyridone adduct is observed. See SI†.
- For palladium C–H ActivationKIE values, see
(a) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS
- For Rhodium(III) C–H activation values, see ref. 11d and, N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13 Search PubMed
- See SI†.
- For information regarding CMD events see previous work which indicates the necessity for a carboxylate source, consistent with a CMD mechanism., D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 Search PubMed
-
(a) L. Li, Y. Jian, W. W. Brennessel and W. D. Jones, Organometallics, 2010, 29, 4593 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information, compound characterizatio, starting material synthesis and kinetic studies. See DOI: 10.1039/c1sc00235j |
| This journal is © The Royal Society of Chemistry 2011 |