Large scale restructuring of porous Pt-Ni nanoparticle tubes for methanol oxidation: A highly reactive, stable, and restorable fuel cell catalyst†
Chun-Hua
Cui
a,
Hui-Hui
Li
b and
Shu-Hong
Yu
*ab
aDivision of Nanomaterials & Chemistry, Hefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: shyu@ustc.edu.cn
bDepartment of Chemistry, University of Science and Technology of China, Hefei, 230026, P. R. China
First published on 18th May 2011
Abstract
We report a large scale restructuring of porous Pt-Ni nanoparticle tubes for electrocatalytic oxidation of methanol, showing high catalytic activity, stability and resistance to poisoning. The surface restructuring highly improved the electrochemical active surface area (ECSA) by potential cycling in a strong acid electrolyte at room temperature. After a long-time stability test, the ECSA can be restored to its initial value after another potential cycling, thus this kind of electrocatalyst shows the potential possibility for next-generation highly restorable catalysts in direct methanol fuel cells.
The design of new catalysts for polymer electrolyte membrane fuel cells (PEMFC) must be considered by a reduction in the amount of noble metals in catalysts.1 One promising alternative is to improve the electrochemical active surface area (ECSA) per mass that involves decreasing the size of particles2 and forming noble-metal shells with inexpensive metal or alloy in the core region.3 However, decreasing the particle size to several nanometres is limited by the conventional synthesis methods using a mass of surfactant or dendrimers as a templates,2a which are barriers for confining the catalytic activities. To increase the surface roughness and active sites, the surface segregation and restructuring may be considered as encouraging routes and are of paramount importance in the quest to investigate the surface rebuilding of catalyst particles, especially large particles, for the enhancement of specific surface area per mass.4
Herein, we report a large scale restructuring of porous Pt-Ni nanoparticle tubes for electrocatalytic oxidation of methanol. The rearranged surface with significant enhancement of ECSA on a large Pt-Ni particle shows high activity, long-term durability and restorable properties for methanol oxidation. This catalyst with porous tubular structure was synthesized by thermal annealing of the anodic aluminum oxide (AAO) template-supported Pt-Ni nanoparticle tubes, which were prepared by a newly developed nonaqueous solution electrochemical method.‡5
The porous Pt-Ni nanoparticle tubes (PNPTs) with a diameter of about 300 nm and 30 at.% Pt were obtained by thermal annealing at 600 °C in reductive conditions. As shown in Fig. 1a, the transmission electron microscopy (TEM) image demonstrates a tubular and porous structure. The tube wall is very thin and the average size of the particles is about 30 nm with a wide size distribution. The tube orifice and the panoramagram can be observed in Fig. S1a and 1b,† and lengths of these tubes are about from one to several micron. These structures may be used as supportless catalysts, and both the outer and inter surface active sites can be exposed for active molecules.6Fig. 2 shows the X-ray diffraction pattern of PNPTs and well-defined peaks corresponding to (111), (200) and (220) planes without any impurity peaks and monocomponent Ni or Pt peaks, indicating the formation of face-centered cubic disordered alloys.7 According to the binary alloy phase diagrams,8 the Pt-Ni system is apt to form ordered alloys with a Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt molar ratio of 1:1 and the formation conditions of ordered Ni3Pt alloys are rigorous. Thus, here the ordered alloys are difficult to be synthesized even when the temperature was increased to 600 °C. In this case, the alloy particle may also demonstrate high mixing and compressive lattice strain. This lattice stress may be considered as one of the strong driving forces for surface reconstruction and rearrangement of surface atoms due to the relaxation of surface free energy and bond energy at the metal-electrolyte interface.9
:Pt molar ratio of 1:1 and the formation conditions of ordered Ni3Pt alloys are rigorous. Thus, here the ordered alloys are difficult to be synthesized even when the temperature was increased to 600 °C. In this case, the alloy particle may also demonstrate high mixing and compressive lattice strain. This lattice stress may be considered as one of the strong driving forces for surface reconstruction and rearrangement of surface atoms due to the relaxation of surface free energy and bond energy at the metal-electrolyte interface.9
 | ||
| Fig. 1 TEM images of PNPT (a) before and (c) after 250 potential cycles. (b) Cyclic voltammogram evolution of PNPT catalyst from 1st to 250th potential cycles. (d) Component ratio change of Pt before and after potential cycling in the bulk by ICP and EDX and near surface region by XPS. | ||
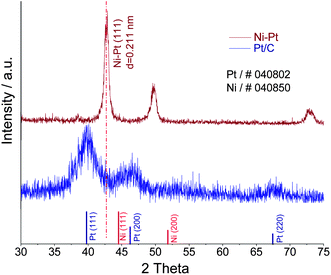 | ||
| Fig. 2 XRD patterns of the PNPTs before potential cycling and the commercial Pt/C. | ||
Electrochemical cyclic voltammetry is a surface-sensitive technique for the determination of the surface oxidation state of the atoms depending on the applied potential.10 Under electrochemical environments, the anodic current peaks of non-noble metals should be detected on the anodic portion of the initial first scan even if certain contaminants are adsorbed on the alloy surface.11 As demonstrated in Fig. 1b, the cyclic voltammograms (CVs) of the initial first and the following potential cycling recorded show the absence of redox peaks of Ni, which should be the qualitative information of the Pt-rich shell formation after thermal annealing depending on the heat of segregation and the surface mixing energy.4a However, the underpotentionally deposited hydrogen (Hupd) between 0.05 and 0.4 V (vs. RHE) can not even be observed, the reasons of which may be due to the few Pt surface sites because of the large particle size and the contaminants. But in surprise, after 250 potential cycles as shown in Fig. 1b, the Hupd and the adsorption/desorption of the oxygenated species above 0.6 V are highly increased, implying the enhancement of the ECSA. Strasser et al.11 demonstrated the well-grounded results that the selective dissolution of non-noble metal on a Pt-contained alloy surface could modify the surface electrocatalytic reactivity resulting from undercoordinated surface sites (surface roughness) and lattice strain. In the dealloying process the components between non-noble metal and Pt will suffer immense change due to the massive leaching of non-noble metal on the alloy surface under potential cycling. However, in this process, no redox peaks of Ni have been obtained and there is a ∼100-fold ECSA increase compared to that of a clean surface after several potential cycles. The enhancement of the ECSA should attribute to the surface roughness. As shown in Fig. 1c, the surface roughness evolution was qualitatively characterized by the change of the Hupd regions. The electron diffraction (ED) pattern after electrochemical treatment becomes slightly continuous, implying the configuration change on the particle surface. After potential cycling, the atomic percentage of Ni changed from ∼70 to 57 at.% Ni. The decrease of metal Ni component should be the persistent surface segregation and concomitant continuous dissolution of Ni.10 The surface segregation induced leaching of non-noble metal in acid electrolyte is a common phenomenon. Wang and coworkers reported that after dealloying the final Ni concentration about 11–27 at.% Ni remains in the PtNi/C catalyst and the dissolution of Ni is highly dependent on the initial alloy composition.3f The results indicate that the Ni atoms will migrate to the surface due to the surface segregation when the potential is cycled in typical fuel cell application ranges. In this experiment, after violent potential cycling there is still about 57 at.% Ni in the Pt-shell Pt-Ni alloy particles although surface segregation results in the dissolution of Ni at the same time of surface restructuring. The experimental identification of the restructuring and surface segregation phenomena will be simultaneously demonstrated in a separate paper. The near surface Pt concentration measured by XPS is similar with that (average Pt content) of the bulk Pt-Ni particles by inductively coupled plasma mass spectrometry (ICP-MS) and energy-dispersive X-ray spectroscopy (EDX). Similar results were also obtained for PtNi3 dealloyed catalysts by EDX line-profile analysis crossing individual particles.3f The component change is highly related to the bulk component ratio, the particle size and even the external environment. The components of the relatively larger particles about 100 nm have been also studied in Fig. S2 and S3.† The selected EDX data indicate that the larger particle with lower at.% Pt has a similar component change with that of smaller particles after potential cycling. High resolution TEM (HRTEM) images of the PNPTs before and after potential cycling are shown in Fig. 3. A lattice spacing of ca. 0.211 nm of the particle crystals corresponds to the lattice spacing of the (111) planes in the fcc Pt-Ni, and obvious lattice spacing widening is not observed in the bulk after potential cycling.
 | ||
| Fig. 3 HRTEM images of the PNPTs (a and b) before and (c and d) after 250 potential cycles between 0.05 and 1.2 V (vs. RHE) in Ar-saturated 0.1 HClO4 solutions with the sweep rate of 250 mV s−1. | ||
The electrocatalytic activity of the PNPTs towards methanol oxidation against a commercial Pt/C catalyst was investigated. The dispersed PNPT solution was loaded onto a glassy carbon electrode and dried at room temperature. Then the potential cycling as depicted in Fig. 1b was performed in Ar-saturated 0.1 M HClO4 solutions at room temperature. After 250 potential cycles, the fresh and highly active surface sites were obtained and the enhanced ECSA was also achieved. The real surface area can be obtained by calculating the areas of H adsorption/desorption after the deduction of the double layer region on the CV. The catalytic activity for methanol oxidation was normalized by this estimated surface area. The CVs of PNPT and commercial Pt/C catalyst in 0.1 M HClO4 and 1.0 M MeOH solutions at a scan rate of 50 mV s−1 are shown in Fig. 4a. The inset shows the corresponding CVs for the same sample in Ar-saturated 0.1 M HClO4 without methanol. The area specific current density per mass was 425 mA cm−2 mg−1Pt at 0.8 V for the PNPT catalyst and double that for the commercial Pt/C catalyst (210 mA cm−2 mg−1). Park et al. reported that the Ni oxides can serve as the oxygen donors and change the electronic structure of the Pt in the Pt-Ni alloys for the enhancement of methanol oxidation.12 After potential cycling, the Pt-shell may form due to the depletion of surface Ni and inhibit the formation of Ni oxides (see Fig. S5†). Therefore, the rearrangement of the surface Pt atoms may built highly active sites. For the methanol oxidation, the poisoning tolerance, If/Ib, can be defined as the peak current ratio of the forward scan (If) to backward scan (Ib).13 The If/Ib of the PNPT catalyst is 1.26, which is higher than that of Pt/C (0.92) and even also higher than that of the porous Pt-Co nanowire catalyst at the maximum value (1.20).12 This high enhancement in If/Ib for the PNPT catalyst indicates that it is both highly active and highly resistant to the poisoning of carbonaceous species. Here, the compressive strain may play a major role in the resistance to the poisoning of carbonaceous species because higher lattice strain can reduce the bond energy of the intermediate oxygenated adsorbates and favor the removal of the carbonaceous species.3d The higher catalytic activity for methanol oxidation was further confirmed by the chronoamperometric experiment at 0.8 V in Fig. 4b. It is obvious that the Pt/C catalyst decays with time and shortly reaches a steady state, while the PNPT catalyst takes a longer time. The higher steady state current of the PNPT catalyst suggest a better performance.
 | ||
| Fig. 4 (a) Cyclic voltammetric curves of Pt/C (dash dot line) and PNPT (straight line) catalysts for methanol oxidation in 0.1 M HClO4 + 1.0 M MeOH solution. The specific currents are normalized to the real Pt surface areas per mass. Inset shows voltammograms of the corresponding electrodes in 0.1 M HClO4 at 50 mV s−1 without MeOH. (b) Chronoamperograms for methanol oxidation at 0.8 V on Pt/C (dash dot line) and PNPT (straight line) in 0.1 M HClO4 + 1.0 M MeOH solution. | ||
The further possible degradation for both Pt/C and PNPT catalysts has been investigated. As we know, the commercial Pt/C catalyst degradation should be due to the Pt particle ripening, agglomeration, dissolution/redeposition and corrosion of carbon support caused decrease of ECSA in hostile electrochemical environments.14 According to the recent research results, after 1000 potential cycles the ECSA of the highly stable free-standing Pt-nanowire membrane decreased by 10%, while the Pt black and Pt/C lost 35% and 70% of the initial ECSAs, respectively.15 However, the degradation of the PNPT catalysts may be different and should be considered as the removal of reconstruction by the adsorbates (in Fig. S4†).16 After the per 2000 s stability test, we performed another potential cycling in Ar-saturated 0.1 M HClO4 solution. The initial several potential cycles can remove the surface adsorption of carbonaceous species and the following potential cycling may induce the restoration of surface microstructures in Fig. S4a-c,† and finally recover and even somewhat further increase the ECSA due to the surface restructuring in Fig. 5. These characteristics may inspire us to design next-generation restorable fuel cell catalysts.
 | ||
| Fig. 5 ECSA % of the PNPTs calculated after each 250 potential cycles in Ar-saturated 0.1 M HClO4 solution. After the 1st, 2nd and 3rd 250 potential cycles, the chronoamperometry (2000 s) was conducted for methanol oxidation at 0.8 V in 0.1 M HClO4 + 1.0 M MeOH solution, respectively. The ECSA calculated with respect to that of the PNPTs after the 1st 250 potential cycles. | ||
In summary, we have designed a large-scale restructuring method to highly improve the ECSA of a Pt-based PNPT catalyst. This kind of catalyst is highly active, and highly resistant to the poisoning of carbonaceous species and shows high stability. Moreover, after potential cycling, the ECSA can be restored to its initial value, indicating that it can be used as a restorable fuel cell catalyst. The results suggest that the restructuring may be considered as a new view to understand the catalytic activity and stability.
Acknowledgements
S. H. Yu acknowledges the special funding support from the National Basic Research Program of China (2010CB934700), the National Natural Science Foundation of China (NSFC, Nos. 91022032, 50732006), and International Science & Technology Cooperation Program of China (2010DFA41170).Notes and references
-
(a) V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS
; (b) J. X. Wang, H. Inada, L. J. Wu, Y. M. Zhu, Y. M. Choi, P. Liu, W. P. Zhou and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 17298 CrossRef CAS
-
(a) K. Yamamoto, T. Imaoka, W. J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397 Search PubMed
-
(a) V. Mazumder, M. Chi, K. L. More and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 9368 Search PubMed
-
(a) K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Angew. Chem., Int. Ed., 2009, 48, 3529 CrossRef CAS
-
(a) C.-H. Cui, H.-H. Li, J.-W. Yu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2010, 49, 9149 Search PubMed
-
(a) S. M. Alia, G. Zhang, D. Kisailus, D. Li, S. Gu, K. Jensen and Y. Yan, Adv. Funct. Mater., 2010, 20, 3742 CrossRef CAS
- T. Ghosh, B. M. Leonard, Q. Zhou and F. J. DiSalvo, Chem. Mater., 2010, 22, 2190 CrossRef CAS
-
P. R. L. Subramanian, D. E. Laughlin in Binary Alloy Phase Diagrams 2nd edn, ed. T. B. Massalski, ASM International, vol. 2, 1990, pp. 1460–1462 Search PubMed
- W. Haiss, Rep. Prog. Phys., 2001, 64, 591 CrossRef CAS
- K. J. J. Mayrhofer, K. Hartl, V. Juhart and M. Arenz, J. Am. Chem. Soc., 2009, 131, 16348 CrossRef CAS
- P. Strasser, S. Koha and J. Greeley, Phys. Chem. Chem. Phys., 2008, 10, 3670 RSC
- K.-W. Park, J.-H. Choi, B.-K. Kwon, S.-A. Lee, Y.-E. Sung, H.-Y. Ha, S.-A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869 CrossRef CAS
- L. Liu, E. Pippel, R. Scholz and U. Gösele, Nano Lett., 2009, 9, 4352 CrossRef CAS
- Z.-B. Wang, P.-J. Zuo, Y.-Y. Chu, Y.-Y. Shao and G.-P. Yin, Int. J. Hydrogen Energy, 2009, 34, 4387 Search PubMed
- H.-W. Liang, X. Cao, F. Zhou, C. H. Cui, W. J. Zhang and S. H. Yu, Adv. Mater., 2011, 23, 1467 Search PubMed
- G. A. Somorjai and M. A. Van Hove, Prog. Surf. Sci., 1989, 30, 201 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S4 contain TEM, EDX, XPS and electrochemical data. See DOI: 10.1039/c1sc00233c |
| ‡ Experimental Section. Chemicals.NaNO3, NiSO4·6H2O, Dimethyl sulfoxide (99% wt.%), C2H5OH (≥99.7% wt.%), HClO4 (70∼72 wt.%) and NaOH were commercially available from Shanghai Chemical Reagent Co. Ltd, Nafion (wt.5%) from Sigma-Aldrich and PtCl2 from J&K chemical Ltd. All the chemical reagents were analytical grade, and used as received without further purification. Synthesis of Porous Pt-Ni Nanoparticle Tubes. The commercial anodic aluminum oxide template (Anodisc 47, Whatman Co., UK, 300 nm in diameter measured by SEM) sputtered with thin Au layer (40 nm thickness) to form an annular base electrode at the bottom of the pores. This thin Au layer was used as the working electrodes in the subsequent electrodeposition (IM6ex electrochemical workstation, Zahner, Germany). For Pt-Ni synthesis, the experiments were performed potentiostatically at −1.2 V in DMSO solvents mixed with 10 mM PtCl2, 30 mM NiSO4·6H2O and 100 mM NaNO3. All electrode potentials were measured relative to an Ag/AgCl (3 M) reference electrode using a Pt foil as a counter electrode. After electrodeposition, the thin Au layer on one side of the AAO template has been erased completely by alumina particles, and washed with doubly deionized H2O and ethanol several times and dried for annealing. The Pt-Ni material was loaded on the bottom of a rectangular ceramic boat and annealed to 600 °C (5 °C min−1 heating rate) in a flow tubular furnace (MTI GSL1400X) under a flowing 5% hydrogen atmosphere (Ar balance). And then the products were exposed by immersing in 1.0 M NaOH solution for 1 h to completely remove the AAO template, and followed by washing with doubly deionized H2O and ethanol several times. Characterization. The resultant phase of the as-synthesized Pt-Ni porous tubes was examined by XRD (Cu-K radiation, = 0.154056 nm). X-ray photoemission spectra were recorded on an ESCALAB-MK-II. Energy-dispersive X-ray spectra (EDX) were taken with a JEOJ-2010 transmission electron microscope with an acceleration voltage of 200 kV. SEM images were obtained with a Zeis SupraTM 40 high resolution field emission scanning electron microscope operating at 5 kV. TEM and HRTEM images were obtained using JEOL-2100F TEM. Inductively coupled plasma mass spectrometry (ICP-MS) data were obtained by Optima 7300 DV. Electrochemical measurements. A platinum foil and Ag/AgCl (3 M) were used as the counter and reference electrodes, respectively. All electrode potentials were recorded with respect to the reversible hydrogen electrode (RHE). A glassy carbon electrode (GCE) (PINE, 5 mm diameter, 0.19625 cm2) was polished to a mirror finish (No. 40-6365-006, Gamma Micropolish Alumina, Buehler; No.40-7212, Microcloth, Buehler) and thoroughly cleaned. The preparation method of the working electrode can be found as follows: ethanol suspensions of 1 mg catalyst (recorded with metal Pt) per millilitre with 0.02 wt.% Nafion (diluted from 5 wt.% Nafion, Sigma-Aldrich) were obtained by ultrasonic mixing for about 20 min. The metal catalyst suspension was transferred onto the GCE substrate, leading to the metal Pt loading of 14.4 μg cm−2 for Pt/C catalyst and 29 μg cm−2 for Pt-Ni catalysts, respectively. Finally, the as-prepared catalyst GCD was dried at room temperature. The potential cycling treatment was performed in Ar-saturated 0.1 M HClO4 solution at the sweep rate of 250 mV s−1 for 250 cycles at room temperature. After potential cycling, the sweep rate was changed at 50 mV s−1 from 0.05 to 1.2 V for ECSA measurement. The methanol oxidation reaction was conducted in Ar-saturated 0.1 M HClO4 and 1.0 M MeOH solution at the sweep rate of 50 mV s−1 and the specific current density per mass were studied at 0.8 V vs. RHE. The real surface Pt normalized chronoamperograms for methanol oxidation at 0.8 V on Pt/C and PNPT catalysts in 0.1 M HClO4 and 1.0 M MeOH solution. |
| This journal is © The Royal Society of Chemistry 2011 |