DOI:
10.1039/C1SC00024A
(Edge Article)
Chem. Sci., 2011,
2, 1016-1024
High-contrast Cu(I)-selective fluorescent probes based on synergistic electronic and conformational switching†
Received
15th January 2011
, Accepted 15th February 2011
First published on 17th March 2011
Abstract
The design of fluorescent probes for the detection of redox-active transition metals such as Cu(I/II) is challenging due to potentially interfering metal-induced non-radiative deactivation pathways. By using a ligand architecture with a built-in conformational switch that maximizes the change in donor potential upon metal binding and an electronically decoupled tunable pyrazoline fluorophore as acceptor, we systematically optimized the photoinduced electron transfer (PET) switching behavior of a series of Cu(I)-selective probes and achieved an excellent fluorescence enhancement of greater than 200-fold. Crystal structure analysis combined with NMR solution studies revealed significant conformational changes of the ligand framework upon Cu(I) coordination. The photophysical data are consistent with a kinetically controlled PET reaction involving only the ligand moiety, despite the fact that Cu(I)-mediated reductive quenching would be thermodynamically preferred. The study demonstrates that high-contrast ratios can be achieved even for redox-active metal cations, provided that the metal-initiated quenching pathways are kinetically unfavorable.
Introduction
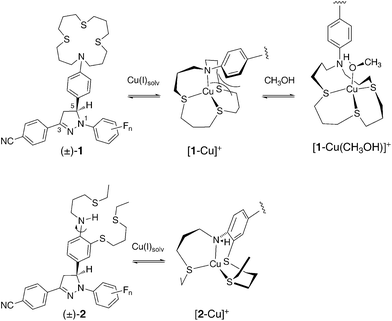
Cation-responsive fluorescent probes have found widespread applications in analytical chemistry, environmental chemistry, materials sciences, and the biological sciences. Fluorescence-based detection technologies are cost-effective and combine excellent sensitivities with a large dynamic range. While many fluorescent probes have been developed for non-redox active metal cations, the detection of metals with readily accessible multiple oxidation states, such as Cu(II/I)1,2 or Fe(III/II)3 poses significant challenges due to interfering quenching pathways. These problems have been addressed through the use of a rigid probe architecture that spatially separates the metal cation-binding site from the fluorophore and a photoinduced electron transfer switching (PET) mechanism that controls the emission intensity in an analyte-dependent manner.4 Interestingly, Cu(I)-responsive PET probes described in the literature consistently display incomplete recovery of the emission quantum yield compared to the corresponding isolated fluorophores.2 Taking into consideration the inherently low redox potential of Cu(I)-complexes,5 it would be reasonable to assume that the compromised fluorescence recovery is due to a Cu(I)-mediated PET quenching pathway. Nevertheless, recent photophysical studies in our laboratory demonstrated that formation of ternary complexes with solvent molecules is the reason behind the less than optimal fluorescence recovery, rather than the redox activity of the metal cation.6 The model system 1 used for these studies was comprised of a 1,3-diaryl-substituted pyrazoline fluorophore, an established platform for the design of PET probes,7,8 and an electronically decoupled thiaza crown receptor for selective binding of Cu(I). As depicted in Scheme 1, binding of Cu(I) to the crown moiety of 1 induces a conformational change, which leads to sterically unfavorable interactions between the ortho-hydrogen atoms of the aniline ring and the thiaza crown backbone. Presumably due to these interactions, complex [1-Cu]+ further equilibrates with solvent molecules to form sterically less encumbered ternary complexes such as [1-Cu(CH3OH)]+, as evidenced by spectroscopic and computational studies.6 Because coordination of a solvent molecule mitigates not only the unfavorable steric interactions, but also weakens binding of Cu(I) to the aniline nitrogen, the electron-donating ability of the aniline moiety is increased which results in a more favorable PET process and thus an incomplete fluorescence recovery. We reasoned that integration of the aniline ring into the thiaza backbone should remedy this problem, an approach that led to the design of pyrazoline probes of type 2 described in this report (Scheme 1).
 |
| | Scheme 1 | |
The receptor moiety of 2 offers an NS3 coordination environment, similar to that of 1, albeit with an acyclic ligand topology for better synthetic accessibility. As demonstrated by an examination of a large number of copper complexes, this change in topology has statistically insignificant effects on the Cu(II/I) redox potentials.9 Funtionalized with only a single thioether substituent, the secondary aniline nitrogen of 2 can readily adopt a trigonal planar geometry for efficient π-donation, which is particularly important for stabilizing the transient radical cation formed in the PET reaction. Upon coordination of Cu(I), the ligand undergoes a conformational change that abrogates the π-donating ability of the aniline nitrogen. Due to the reduced donor strength of the aniline moiety, PET quenching should be effectively suppressed and hence yield a high fluorescence contrast. In addition to redesigning the receptor unit, we also took advantage of the inherent electronic tunability of the pyrazoline fluorophore to optimize the fluorescence contrast upon Cu(I)-binding as previously demonstrated.8,10,11
Results and discussion
Synthesis
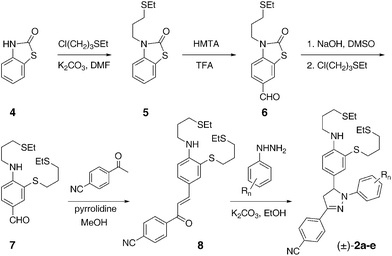
The target compounds 2a–e were assembled in six synthetic steps (Scheme 2). Incorporation of the mono-alkylated aniline moiety posed some challenges, since direct alkylation was expected to yield a mixture of the mono- and dialkylated products, and the mono-alkylated aniline nitrogen would promote undesired side reactions in the subsequent formylation under standard Vilsmeier–Haack conditions. While masking of the unused valency on the aniline nitrogen could remedy these problems, common protective groups such as alkylcarbamates or arylsulfonamides are sterically too demanding, thus enforcing a non-planar geometry on the aniline nitrogen due to the presence of the ortho-positioned thioether substituent. This in turn would abolish nitrogen π-donation and substantially reduce the nucleophilicity of the aniline ring towards formylation.12 To address these issues, we devised a synthesis starting from benzothiazolinone 4. The carbonyl bridge in this compound not only protects the free valency of the aniline nitrogen, but simultaneously lowers its pKa for efficient mono-alkylation. The resulting intermediate 5 was sufficiently nucleophilic to undergo regioselective formylation in the 6-position of the benzothiazolinone to give 6 with 87% yield. Hydrolysis of the carbonyl protective group released the sulfhydryl functionality, which was directly alkylated without isolating the air-sensitive thiophenol intermediate. The resulting benzaldehyde derivative 7 was converted to the chalcone 8, and condensation with the corresponding fluoro-substituted phenylhydrazine derivatives concluded the synthesis of the racemic pyrazoline probes (±)-2a–e. Because the 1-aryl group of the pyrazoline is introduced in the last step, the fluoro-substituents attached to this ring can be readily varied based on a single precursor 8. For the photophysical characterization, we additionally synthesized the pyrazoline fluorophores 3b–c as reference compounds, in which the aniline donor moiety was replaced with an unsubstituted phenyl ring.
 |
| | Scheme 2 | |
Photophysical characterization
The photophysical properties of the pyrazoline probes (±)-2a–e and their fluorescence responses towards Cu(I) were determined in methanol as solvent (Table 1). Estimates for the photoinduced electron transfer driving forces were obtained according to the Rehm-Weller formalism14 and are based on the experimentally determined ground state donor and acceptor potentials of each derivative (see ESI†). Consistent with lowest excited states that have considerable charge-transfer character, compounds (±)-2a–e exhibit broad unstructured absorption and emission bands (Fig. 1).
Table 1 Photophysical data of pyrazoline probes 2a–e and reference compounds 3b–c in methanol at 298 K
| Compd |
Rn |
abs λmax (nm) |
em λmax (nm) |
Stokes shift (cm−1) |
ΔE00a (eV) |
ΔGetb (eV) |
ΦFc |
f
e
d
|
| Neutrale |
Cu(I)
f |
|
Zero-zero transition energy in methanol; estimated as the average of the peak absorption and emission energies.
Photoinduced electron transfer free energy ΔGet estimated based on the Rehm-Weller formalism14 using experimental ground-state donor and acceptor potentials in CH3CN (see ESI†).
Fluorescence quantum yield in methanol; quinine sulfate as reference.13
Fluorescence enhancement factor fe = ΦF/Φneutral.
Neat methanol.
10 μM [Cu(I)(CH3CN)4]PF6 and 5 μM probe in methanol (0.1% acetonitrile).
Signal to noise ratio insufficient for reliable determination.
|
|
2a
|
3-F
|
391 |
486 |
5,000 |
2.86 |
−0.18 |
0.017 |
0.57 |
34 |
|
2b
|
2,5-F2 |
380 |
479 |
5,440 |
2.93 |
–0.27 |
0.007 |
0.54 |
74 |
|
2c
|
2,3,5-F3 |
371 |
463 |
5,360 |
3.01 |
−0.36 |
0.002 |
0.49 |
210 |
|
2d
|
2,3,5,6-F4 |
354 |
441 |
5,570 |
3.16 |
−0.46 |
0.001 |
0.24 |
160 |
|
2e
|
2,3,4,5,6-F5 |
346 |
432 |
5,750 |
3.23 |
−0.55 |
n.d.g |
0.21 |
— |
|
3b
|
2,5-F2 |
376 |
482 |
5,840 |
2.93 |
— |
0.54 |
— |
— |
|
3c
|
2,3,5-F3 |
370 |
467 |
5,600 |
3.00 |
— |
0.64 |
— |
— |
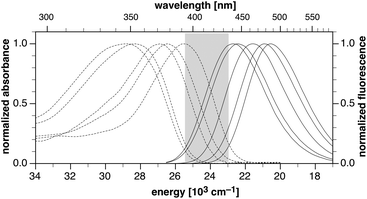 |
| | Fig. 1 Normalized absorption (dotted traces) and emission spectra (solid traces) of pyrazoline probes 2a–e in methanol. The shaded area indicates the tunable range of the excited-state energy ΔE00. | |
With increasing number of fluoro substituents on the 1-aryl ring, the absorption and emission maxima gradually shifted to higher energies, and consequently, the zero-zero excited state energies ΔE00 increased stepwise in increments of approximately 0.1 eV from 2.86 to 3.23 eV. The observed trend indicates a gradually decreasing charge delocalization from the 1-N-pyrazoline nitrogen to the electron-deficient 3-aryl ring, in both the ground and excited state of the fluorophore.8,10,11 Since the acceptor potential of 1,3,5-triarylpyrazolines is not significantly affected by substituents in the 1-position of the pyrazoline core,8,10,11 the PET driving forces similarly increased in a stepwise fashion from −ΔGet = 0.18 to 0.55 eV. For all derivatives, PET is strongly exergonic which results in efficient quenching of the fluorescence emission. Concomitant with the increasingly favorable PET driving forces, the quantum yields gradually decreased from 1.7% to 0.1% (Table 1).
Fluorescence response and selectivity towards metal cations
Upon addition of Cu(I) from a stock solution of [Cu(I)(CH3CN)4]PF6 in methanol (stabilized with 10% acetonitrile), all derivatives 2a–e displayed strong fluorescence increases. The emission intensity reached saturation at an equimolar concentration of Cu(I), indicating formation of a complex with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal–ligand stoichiometry (vide infra). The quantum yields of the Cu(I)-bound species followed the same trend as observed in methanol alone, where the 3-fluoro substituted derivative 2a showed the highest degree of fluorescence recovery and the perfluorophenyl derivative 2e the lowest. In agreement with the previously devised model,10 the associated fluorescence enhancement factors fe, defined as the ratio of the quantum yields in the presence and absence of Cu(I), showed a bell-shaped distribution across the compound series. The highest contrast ratio of greater than 200:1 was achieved with the trifluoro-substituted derivative 2c, which offers an intermediate PET driving force of −ΔGet = 0.36 eV. Although the smaller PET driving forces of the mono- and difluoro-substituted derivatives 2a and 2b led to higher quantum yields upon saturation with Cu(I), the corresponding enhancement factors were significantly lower. In these derivatives, the recovered quantum yields are presumably limited by the intrinsic fluorescence properties of the pyrazoline fluorophores rather than PET quenching. The difluoro-substituted pyrazoline derivative103b, which contains an unsubstituted phenyl ring instead of the electron rich chelating moiety, displayed essentially the same quantum yield (ΦF = 0.54) as the corresponding Cu(I)-coordinated derivative 2b, thus confirming this hypothesis. In contrast, the 5-phenyl substituted reference compound 3c exhibited a markedly higher quantum yield of ΦF = 0.64 compared to Cu(I)-saturated 2c, indicating either the presence of ternary complexes with solvent molecules, or an insufficient increase of the donor potential to fully abrogate the competing non-radiative PET deactivation pathway. Following the same trend, further increased PET driving forces as in derivatives 2d and 2e resulted in not only reduced contrast ratios but also further diminished quantum yields.
:1 metal–ligand stoichiometry (vide infra). The quantum yields of the Cu(I)-bound species followed the same trend as observed in methanol alone, where the 3-fluoro substituted derivative 2a showed the highest degree of fluorescence recovery and the perfluorophenyl derivative 2e the lowest. In agreement with the previously devised model,10 the associated fluorescence enhancement factors fe, defined as the ratio of the quantum yields in the presence and absence of Cu(I), showed a bell-shaped distribution across the compound series. The highest contrast ratio of greater than 200:1 was achieved with the trifluoro-substituted derivative 2c, which offers an intermediate PET driving force of −ΔGet = 0.36 eV. Although the smaller PET driving forces of the mono- and difluoro-substituted derivatives 2a and 2b led to higher quantum yields upon saturation with Cu(I), the corresponding enhancement factors were significantly lower. In these derivatives, the recovered quantum yields are presumably limited by the intrinsic fluorescence properties of the pyrazoline fluorophores rather than PET quenching. The difluoro-substituted pyrazoline derivative103b, which contains an unsubstituted phenyl ring instead of the electron rich chelating moiety, displayed essentially the same quantum yield (ΦF = 0.54) as the corresponding Cu(I)-coordinated derivative 2b, thus confirming this hypothesis. In contrast, the 5-phenyl substituted reference compound 3c exhibited a markedly higher quantum yield of ΦF = 0.64 compared to Cu(I)-saturated 2c, indicating either the presence of ternary complexes with solvent molecules, or an insufficient increase of the donor potential to fully abrogate the competing non-radiative PET deactivation pathway. Following the same trend, further increased PET driving forces as in derivatives 2d and 2e resulted in not only reduced contrast ratios but also further diminished quantum yields.
In view of the excellent fluorescence contrast offered by derivative 2c, the following solution chemistry studies focused on this compound alone. We first investigated the Cu(I)-complexation equilibrium by means of a fluorescence titration. To a solution of 2c in methanol were added 0.1 molar equivalent aliquots of Cu(I), and upon equilibration, emission spectra were recorded with excitation at 362 nm (Fig. 2A). Consistent with a PET fluorescence switching mechanism, the peak wavelength of each spectrum remained constant throughout the entire titration. A molar-ratio plot of the fluorescence intensity at 465 nm revealed a linear increase up to an equimolar concentration of Cu(I), indicating a 1:1 metal–ligand stoichiometry and strong binding with near quantitative complex formation at each titration point (Fig. 2A, inset). Because there is a large error associated with fitting data from binding isotherms involving greater than 80% complex formation,15 the above titration data were not suitable for extracting a reliable complex stability constant.
![Fluorescence response of pyrazoline probe 2c towards monovalent copper in MeOH at 298 K. A) Fluorescence titration of 2c (5 μM) with [Cu(i)(CH3CN)4]PF6 (excitation at 362 nm). Inset: Molar-ratio plot of the fluorescence intensity at 465 nm. B) Fluorescence response of 2c as a function of various metal cations (excitation at 370 nm, emission at 463 nm). Black bars: equimolar concentration of 2c and the indicated metal cation. Grey bars: Competition with equimolar amounts of [Cu(i)(CH3CN)4]PF6 and the respective metal cation. C) Time-resolved fluorescence decay profile of 2c saturated with Cu(i) ([Cu(i)(CH3CN)4]PF6; IRF = instrument response function, excitation at 372 nm, fluorescence emission acquired at 460 nm).](/image/article/2011/SC/c1sc00024a/c1sc00024a-f2.gif) |
| | Fig. 2 Fluorescence response of pyrazoline probe 2c towards monovalent copper in MeOH at 298 K. A) Fluorescence titration of 2c (5 μM) with [Cu(I)(CH3CN)4]PF6 (excitation at 362 nm). Inset: Molar-ratio plot of the fluorescence intensity at 465 nm. B) Fluorescence response of 2c as a function of various metal cations (excitation at 370 nm, emission at 463 nm). Black bars: equimolar concentration of 2c and the indicated metal cation. Grey bars: Competition with equimolar amounts of [Cu(I)(CH3CN)4]PF6 and the respective metal cation. C) Time-resolved fluorescence decay profile of 2c saturated with Cu(I) ([Cu(I)(CH3CN)4]PF6; IRF = instrument response function, excitation at 372 nm, fluorescence emission acquired at 460 nm). | |
We further tested the fluorescence response of 2c towards a range of other metal cations. As illustrated with the bar-graph in Fig. 2B, only coordination of Cu(II) led to an appreciable increase of the fluorescence intensity, albeit significantly smaller compared with the enhancement induced by Cu(I). This fluorescence increase was instantaneously reversed by addition of a stoichiometric amount of 1,4,8,11-tetrathiacyclotetradecane ([14]aneS4), a macrocylic chelator that has a 107-fold higher affinity towards Cu(I) compared to Cu(II) (Kd (Cu2+) = 4.3).16 This observation indicates that the Cu(II)-mediated fluorescence increase is in fact due to binding of Cu(I), presumably formed through spontaneous autoreduction and concomitant oxidation of the solvent.17 Competition experiments with equimolar amounts of Cu(I) and each of the metal cations consistently showed full recovery of the fluorescence emission, indicating a good selectivity towards Cu(I) over all tested metal cations.
While the molar-ratio titration data established the stoichiometry of the major complex formed in solution, the method is not suitable to assess whether the formation of ternary complexes might compromise the Cu(I)-promoted fluorescence recovery of derivatives 2c, as such species would not alter the overall metal-probe stoichiometry. Previous studies demonstrated that the kinetics for the interconversion of such coordination species is fast with regard to the NMR time scale, but sufficiently slow to be captured by pico-second time resolved fluorescence spectroscopy.10 The fluorescence decay profile of 2c in the presence of an equimolar amount of Cu(I) fitted well to a monoexponential decay model with a fluorescence lifetime of 2.80 ± 0.01 ns (χ2 = 1.324, Fig. 2C), thus supporting the formation of a single coordination complex without involvement of additional species. For comparison, the reference compound 3c lacking the metal-chelating moiety also exhibited a monoexponential decay profile, though with a somewhat longer fluorescence lifetime of 3.70 ± 0.001 ns (χ2 = 1.140, Fig. S3, ESI†). Given the absence of other coordination species, the difference in lifetimes might be attributed to a competing PET quenching pathway that is still active in the presence of Cu(I), albeit much slower compared to the free probe. Considering the fluorescence quantum yields of 0.49 and 0.64 for Cu(I)-bound 2c and reference compound 3c, we obtained the radiative deactivation rates kr = ΦF/τF of 1.74 × 108s−1 and 1.73 × 108s−1, respectively. The nearly identical values indicate that emission occurs from excited states with closely related properties.
To evaluate the redox properties of the Cu(II/I) couple coordinated to the acyclic thiazaether receptor of 2c, we conducted slow-scan cyclic voltammetry studies in methanol using tetrabutylammonium hexafluorophosphate as the electrolyte.16 A solution of 2c containing an equimolar amount of [Cu(I)(CH3CN)4]PF6 showed a new redox process with a formal half-wave potential of E1/2 = 0.02 V vs. Fc+/0, which was substantially lower compared to the potential of 0.54 V observed for oxidation of the aniline moiety of 2c (Fig. S2, ESI†). At a scan rate of 50 mV s−1, the measured peak-to-peak separation of ΔEp = 90 mV was significantly higher than the theoretical value of 59 mV for a one-electron process, indicating a quasi-reversible electrochemical reaction. At higher scan rates the peak-to-peak separation further increased and reached 130 mV at 500 mV s−1. The observed quasi-reversible electrochemical behavior is typical for copper complexes, which must undergo large structural reorganization in the course of the redox reaction due to the substantially different coordination geometry preferences of Cu(I)versusCu(II).5
Crystallographic structural determination
To characterize the coordination geometry of Cu(I) with the redesigned receptor moiety, we determined the crystal structure of the Cu(I) complex with the truncated ligand 9 lacking the pyrazoline fluorophore (shown in Fig. 4). Crystallographic data are compiled in Table 2, and a list of selected bond distances and bond angles are presented in Table 3. An ORTEP drawing of the complex [Cu(I)-9]+ showing the atom numbering scheme is depicted in Fig. 3. As anticipated, the NS3 donor set places the copper center into a tetrahedral coordination environment; however, there are considerable deviations from a regular tetrahedral geometry. The two six-membered chelate rings adopt a chair conformation with bite angles of N–Cu–S = 107.0° and S–Cu–S = 103.3°. The two chelate planes as defined by S1–Cu1–S2 and N1–Cu1–S3 intersect at an angle of 75.6°, which is considerably less than the ideal tetrahedral dihedral angle of 90°. Furthermore, the five-membered chelate ring exhibits a significantly more acute bite angle of N–Cu–S = 83.3° and places the copper center by 0.999 Å above the benzene ring plane. The Cu–S bond lengths are non-uniform and likely reflect the different length and type of carbon bridges between the donor atoms and the strain associated with accommodating the shorter Cu–N bond. The sum of bond angles at the aniline nitrogen is 332.2°, indicating a pyramidalization that approaches almost the ideal value of 328.5° as expected for an sp3 hybridized center. Consistent with the absence of ternary complex formation, no other close contacts were found in proximity to the copper center.
![ORTEP drawing and atom numbering scheme of the cationic unit in the crystal structure of in [Cu(i)-9]PF6. Ellipsoids shown represent 50% probability. Hydrogen atoms have been omitted for clarity.](/image/article/2011/SC/c1sc00024a/c1sc00024a-f3.gif) |
| | Fig. 3 ORTEP drawing and atom numbering scheme of the cationic unit in the crystal structure of in [Cu(I)-9]PF6. Ellipsoids shown represent 50% probability. Hydrogen atoms have been omitted for clarity. | |
![Dynamic 1H NMR study of the Cu(i)-ligand 9 self exchange equilibrium in CD3OD at 298 K. A) Aromatic region of the 1H NMR spectrum of free ligand 9 (bottom), complex [Cu(i)-9]PF6 (top), and equimolar mixture of the ligand and complex (middle). B) 1H NMR spectra of solutions containing equimolar amounts of ligand 9 and [Cu(i)-9]PF6 at the indicated total ligand concentration. C) Linear correlation of the observed 1H NMR exchange rate (obtained from full line shape analysis with gNMR22) and the total ligand concentration according to eqn (3).](/image/article/2011/SC/c1sc00024a/c1sc00024a-f4.gif) |
| | Fig. 4 Dynamic 1H NMR study of the Cu(I)-ligand 9 self exchange equilibrium in CD3OD at 298 K. A) Aromatic region of the 1H NMR spectrum of free ligand 9 (bottom), complex [Cu(I)-9]PF6 (top), and equimolar mixture of the ligand and complex (middle). B) 1H NMR spectra of solutions containing equimolar amounts of ligand 9 and [Cu(I)-9]PF6 at the indicated total ligand concentration. C) Linear correlation of the observed 1H NMR exchange rate (obtained from full line shape analysis with gNMR22) and the total ligand concentration according to eqn (3). | |
|
final R indices (I > 2σ(I)); indices for all data in parenthesis
|
| Empirical formula |
C16H27CuF6NPS3 |
| Formula weight |
538.08 |
| Crystal size/mm |
0.54 × 0.07 × 0.06 |
| Space group, system |
P21/c, monoclinic |
| Unit cell dimensions |
a = 15.503(3) Å, α = 90° |
|
b = 13.996(3) Å, β = 109.377(3)° |
|
c = 10.645(2) Å, γ = 90° |
|
V/Å3 |
2178.9(8) |
|
Z
|
4 |
|
D
c/g cm−3 |
1.640 |
|
λ/Å |
0.71073 Å |
|
T/K |
173(2) |
|
μ/mm−1 |
1.416 |
|
R(F)a |
0.0795 [0.1615] |
|
R
w(F2)a |
0.2037 [0.2765] |
Table 3 Selected bond distances (Å) and angles (deg) for the cationic unit in [Cu(I)-9]PF6
| Cu(1)–N(1) |
2.107(7) |
N(1)–Cu(1)–S(3) |
107.0(2) |
| Cu(1)–S(1) |
2.360(2) |
S(1)–Cu(1)–S(2) |
103.28(8) |
| Cu(1)–S(2) |
2.247(2) |
S(1)–Cu(1)–S(3) |
115.71(9) |
| Cu(1)–S(3) |
2.225(2) |
S(2)–Cu(1)–S(3) |
127.20(10) |
| |
|
C(2)–N(1)–Cu(1) |
109.0(5) |
| N(1)–Cu(1)–S(1) |
83.3(2) |
C(12)–N(1)–Cu(1) |
111.8(6) |
| N(1)–Cu(1)–S(2) |
111.8(2) |
C(2)–N(1)–C(12) |
111.4(7) |
2D [1H,15N] HSQC NMR experiments
The stability of the radical ion pair intermediate formed in the course of the PET reaction depends on the π-donating ability of the aniline nitrogen, and therefore, the degree of nitrogen pyramidalization upon metal coordination is the most critical structural parameter that determines the switching response of the probe. Because pyramidalization is associated with a change from sp2 to sp3 hybridization, NMR coupling constants may be used as a measure to gauge the magnitude of the geometrical change upon metal binding in solution. Binsch et al. proposed an empirical relationship (1) that relates the amount of s-character of nitrogen bond orbitals directly to experimental 1J(15N–H) coupling constants.18| | | %s = 0.43 × 1J(15N–H) − 6 | (1) |
To determine the 1J(15N–H) values of ligand 9 and its Cu(I) complex in solution, we utilized a 2D [1H,15N] HSQC NMR experiment, which is sufficiently sensitive to acquire data of samples with natural 15N isotope abundance (Table 4).19 The observed 1J(15N–H) value for ligand 9 translates into 33.1% s-character, which agrees well with the expected sp2 hybridization of an uncoordinated aniline nitrogen. Consistent with the pyramidalization observed in the crystal structure, the Cu(I)-complex of 9 revealed a substantially smaller coupling constant corresponding to a reduced s-character of 29.3%. The direct interaction of Cu(I) with the aniline nitrogen is further reflected in the large 15N chemical shift change from −311.7 to −323.0 ppm (Table 4).
| |
Parameter |
Ligand 9 |
Complex [Cu-9]PF6 |
|
CH3NO2 as external reference.
Referenced to TMS.
Spectra provided with the ESI.†
|
| 2D [1H,15N] |
δ (15N)a |
−311.7 |
−323.0 |
|
HSQC
NMR
c |
δ (1H–N)b |
5.388 |
6.090 |
|
J
NH [Hz] |
91.0 |
82.2 |
|
1H-NMR
|
δ (1Ha) |
6.573 |
7.378 |
| δ (1Hb) |
6.666 |
7.413 |
| δ (1Hc) |
7.164 |
7.417 |
| δ (1Hd) |
7.339 |
7.722 |
Dynamic 1H NMR studies
We further characterized the solution equilibrium between ligand 9 and Cu(I) in CD3OD based on dynamic 1H NMR experiments.20,21 As illustrated with Fig. 4, Cu(I) binding resulted in large shifts of the aniline proton resonances. In agreement with pyramidalization of the aniline nitrogen upon Cu(I) binding, the chemical shift changes of the ortho- and para positioned proton resonances (labeled as Ha and Hc in Fig. 4A) were particularly pronounced (Table 4). At an equimolar concentration of ligand 9 and [Cu(I)-9]PF6, only a single set of proton resonances was observed, thus indicating a fast metal exchange equilibrium relative to the NMR time scale. Full line shape analysis using the spectra of the free and fully complexed ligand as reference yielded a pseudo first order exchange rate constant of 16,100 s−1 at 15 mM ligand concentration. In agreement with an associative mechanism, the exchange rate gradually decreased with decreasing ligand concentrations and reached 4,700 s−1 at 3.6 mM (Fig. 4B). The underlying associative exchange can be described by the degenerate self exchange equilibrium in eqn (2),| | 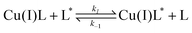 | (2) |
which is related to the observed two site spin exchange system according to eqn (3)where ka and kb refer to the experimental forward and reverse pseudo first order NMR exchange rate constants (see ESI†).21,23 A plot of the observed exchange rate constants versus the total ligand concentration yielded the bimolecular rate constant k1 = 2 × 106s−1M−1 (Fig. 4C). Based on these data, we obtained an average lifetime τ = 1/ka of 200 ms for ligand-bound Cu(I) in an equimolar mixture of complex and ligand at a concentration of 5 μM, which was used for the fluorescence experiments.
The low donor potential of the Cu(I)-thiazaether complex implies that PET mediated quenching of the pyrazoline fluorescence should become more efficient upon binding of Cu(I) to 2c. With this in mind, the strong fluorescence turn-on response of the probe might appear as a contradiction; however, as previously demonstrated for the pyrazoline probes of type 1, the kinetics for PET quenching from Cu(I) is too slow to effectively compete with the rapid excited state deactivation rate of k0 = 1/τF = 2.7 × 108s−1. Not only is the electron transfer process hampered by the high reorganizational barriers associated with all Cu(II/I) complexes,5 but also through the larger distance and thus poorer electronic coupling with the fluorophore π-system. For this reason, PET switching occurs under kinetic control and is dictated by the apparent increase of the donor potential of the aniline moiety upon coordination of Cu(I), irrespective of the thermodynamic stability of the Cu(II/I) couple. As the trigonal planar aniline nitrogen is rotated out of plane upon Cu(I) coordination, the nitrogen lone-pair is no longer available for stabilizing the radical cation formed upon PET. Even in the absence of Cu(I), such a conformational change has a substantial effect on the stability of the aniline radical cation. For example, the reduction potential of ortho substituted tert-butyl-N,N-dimethylaniline is approximately 0.3 V higher compared to the para substituted isomer.24 Because the change in donor potential upon analyte binding is directly related to the electron transfer kinetics,25 we can utilize the experimental data to estimate the apparent potential change that is responsible for the Cu(I)-promoted fluorescence enhancement of 2c. Based on the recovered quantum yield of ΦF′ = 0.49 and the photophysical parameters ΦF and τf of the unquenched reference compound 3c, we can estimate the PET rate constant ket′ in the presence of Cu(I) as| | 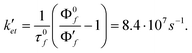 | (4) |
If we assume that the decrease of the electron transfer rate constant ket to ket′ upon Cu(I) binding is solely due to an increase in donor potential ΔE, we can relate ket′ and ΔE according to semiclassical Marcus theory:
| | 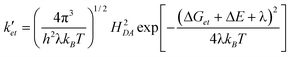 | (5) |
After solving eqn (5) for ΔE we obtain
| | 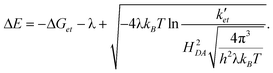 | (6) |
By approximating the reorganization energy and electronic coupling with λ = 0.54 eV and HAD = 18 cm−1, respectively, both of which were previously derived from a series of structurally closely related pyrazoline derivatives,10 we obtain according to eqn (6) an approximate donor potential difference of ΔE = 0.46 V. This estimate hinges on the assumption that the reorganization energy of the PET reaction is not significantly altered in the presence and absence of Cu(I).
In polar solvents such as methanol, λ is dominated by the solvent reorganization contribution λs, therefore, we would expect only minor changes for λ upon Cu(I) coordination, since only the aniline donor, and not the metal ion, participates in the PET process. Similarly, the electronic coupling HDA between the aniline moiety and the pyrazoline fluorophore should not significantly change upon Cu(I)-coordination. In view of the above mentioned potential increase of 0.3 V for the out-of-plane rotation of a dimethylamino group, the estimated value of 0.46 V is not substantially higher, thus supporting the above assumption that the difference in PET reorganization energy between the Cu(I)-bound and free form is small. Altogether, the experimental data offer a comprehensive view of the overall energetics of the Cu(I)-induced electron transfer switching with probe 2c. As illustrated with the simplified Jablonski diagram in Fig. 5, the excited state manifold undergoes considerable changes upon coordination of Cu(I). In the absence of the metal ion, the electron transfer state 1ET lies at 2.65 eV, which is 0.36 eV lower than the S1 energy level of the excited fluorophore (3.01 eV). Hence, the electron transfer reaction is thermodynamically favorable and occurs with a time constant of 11 ps (calculated based on ket = k0(Φ0/Φ − 1), which is more than 300-times faster than the excited state deactivation from S1 in the absence of the donor D, thus resulting in strong fluorescence quenching. As demonstrated by femtosecond transient absorption studies, the intermediately formed radical ion pair [D˙+–A˙−] subsequently undergoes fast charge recombination back to the ground state with a time constant of less than 100 ps.6 Upon coordination of Cu(I), an entirely different picture emerges. The 1ET state now resides 0.1 eV above the energy of the emissive singlet state S1, and the associated time constant is more than 3-times slower compared to excited state deactivation from S1, resulting in efficient radiative deactivation from S1 with a high fluorescence quantum yield. Although electron donation from Cu(I) would yield a highly stabilized radical ion pair [Cu2+–D–A˙−] with an estimated energy of 2.13 eV, the kinetics of this pathway is too slow to compete with radiative deactivation due to the large reorganization energy of the Cu(I/II) couple. For the same reason, charge recombination of the radical ion pair [Cu+–D˙+–A˙−] is very unlikely to proceed via the energetically lower-lying [Cu2+–D–A˙−] species, and therefore occurs through direct back electron transfer as in the absence of Cu(I).
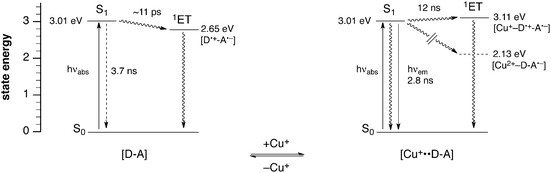 |
| | Fig. 5 Simplified Jablonski diagrams illustrating the energetics of the Cu(I)-dependent photoinduced electron transfer switching of probe 2c (S0 = singlet ground state, S1 = lowest excited singlet state, 1ET = singlet electron transfer state, D = donor, A = acceptor). | |
In our previous model study of a series of 10 pyrazoline derivatives, we derived an optimization map that related the PET driving force −ΔGet and change in donor potential ΔE upon Cu(I) binding with the fluorescence enhancement factor fe.10 As the electronic coupling and reorganization energies are expected to be similar for structurally closely related compounds, the map should also be suitable to rationalize the fluorescence properties of probes 2a–e (Fig. 6A). For example, at a driving force of −0.37 eV estimated for 2c, the contour plot indicates a maximal fluorescence enhancement factor of approximately 200 and a potential difference of 0.46 V.
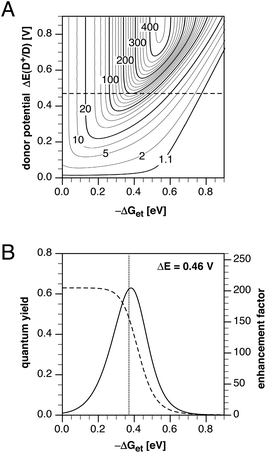 |
| | Fig. 6 A) Contour plot of the fluorescence enhancement factor fe as a function of the PET driving force and change in donor potential ΔE(D+/D) calculated based on equation S6.† For fe between 20 and 420, contour lines are drawn in intervals of 20. Variables: k0 = 2.7 × 108s−1, HDA = 18 cm−1, λ = 0.54 eV, T = 298 K. B) Quantum yield (dashed trace) and fluorescence enhancement factor (solid trace) as a function of the PET driving force for a fixed donor potential difference of ΔE = 0.46 V, calculated based on equations S7 and S9,† respectively. | |
Consistent with the experimental data, compounds with further increased driving forces (2d and 2e) or decreased driving forces (2a and 2b) are expected to yield significantly smaller fluorescence enhancements. To further illustrate this trend, Fig. 6B shows a profile of the contour map across the constant potential value of ΔE = 0.46 V (solid trace). Also in agreement with the experimental data, a plot of the expected quantum yield in the presence of Cu(I)versus the PET driving force shows that the optimal fluorescence enhancement does not coincide with the highest achievable quantum yield (Fig. 6B, dashed trace).
Conclusions
By fully integrating the aniline donor into the backbone of an acyclic NS3 coordination environment, we successfully addressed the adverse effects of ternary complex formation on the fluorescence switching behavior of thiaza crown-based donor–acceptor fluorescent probes. The conformational changes associated with Cu(I)-coordination produced a large increase in donor potential, which was exploited for efficient PET switching. Systematic optimization of the PET parameters by tuning of the photophysical properties of a series of pyrazoline-based probes yielded a greater than 200-fold fluorescence enhancement upon saturation with Cu(I). The photophysical data are consistent with a kinetically controlled PET reaction involving only the aniline donor unit, despite the fact that Cu(I)-mediated reductive quenching is greatly favored thermodynamically. While all probes were characterized in methanol due to insufficient aqueous solubility, we anticipate that the results equally translate to hydrophilic derivatives containing water-solubilizing substituents. Most importantly, the study demonstrates that high-contrast fluorescence probes can be achieved even for redox-active metal cations, providing that the associated quenching pathways cannot kinetically compete with radiative deactivation.
Experimental
Synthesis
Procedures for the synthesis of pyrazoline derivatives 2a–e, 3b–c and all intermediates are provided in the Supplementary Information.†
Sample solutions were filtered through 0.2 μm Teflon membrane filters to remove interfering dust particles or fibers. UV-vis absorption spectra were recorded at 25 °C using a Varian Cary Bio50 UV-vis spectrometer with constant-temperature accessory. Steady-state emission and excitation spectra were recorded with a PTI fluorimeter. For all measurements the path length was 1 cm with a cell volume of 3.0 mL. The fluorescence spectra have been corrected for the spectral response of the detection system (emission correction data provided by instrument manufacturer) and for the spectral irradiance of the excitation channel (via a calibrated photodiode). Fluorescence titrations with Cu(I) were carried out at a fluorophore concentration of 5 μM in MeOH by adding 0.1 molar equivalents from a 1 mM stock solution of [Cu(I)(CH3CN)4]PF6 in MeOH containing 10% CH3CN (v/v). To assess the metal ion selectivity of 2c, the probe was equilibrated with an equimolar amount of the respective metal cation in methanol (5 μM) and the emission spectrum was acquired with excitation at 370 nm. An equimolar amount of [Cu(I)(CH3CN)4]PF6 (1 mM stock solution in MeCN) was subsequently added and the emission spectra acquired again with excitation at 370 nm. Stock solutions of the following metal salts in water were used: Mg(NO3)2·6H2O, Ca(BF4)2·2H2O, MnSO4·2H2O, Fe(BF4)2·6H2O, Ni(NO3)2·6H2O, Cu(TfO)2, Zn(OTf)2, and CdCl2. Quantum yields were determined using quinine sulfate dihydrate in 1.0 N H2SO4 as the fluorescence standard (Φf = 0.54 ± 0.05).13
Time-resolved fluorescence decay data of 2c and 3c were acquired at an emission wavelength of 460 nm with a sample concentration of 0.8 μM in methanol using a single photon counting spectrometer (Edinburgh Instruments, LifeSpec Series) equipped with a pulsed laser diode as the excitation source (372 nm, FWHM = 80 ps, 10 MHz repetition rate, 1024 channel resolution). Sample solutions of the fluorophore saturated with Cu(I) were prepared based on steady-state fluorescence titrations as described above. The time decay data were analyzed by non-linear least squares fitting with deconvolution of the instrumental response function using the FluoFit software package.26
The cyclic voltammograms were acquired in methanol or acetonitrile containing 0.1M Bu4NPF6 as the electrolyte using a CH-Instruments potentiostat (Model 600A). The samples were measured under inert gas at concentrations ranging between 50 μM to 3 mM in a single compartment cell with a glassy carbon working electrode and a Pt counter electrode. An aqueous Ag/AgCl reference electrode (3 M KCl) was used in methanol and a non-aqueous Ag/AgNO3 (10 mM AgNO3 in CH3CN/Ag wire) reference electrode in acetonitrile. The half-wave potentials were referenced to ferrocene as the internal or external standard as recommended by IUPAC.27 Measurements were performed with a scan rate of 100 mV s−1, or as indicated in the text.
2D [1H,15N] HSQC NMR spectra of ligand 9 and [Cu(I)-9]PF6 were acquired on a 500 MHz Bruker DMX NMR spectrometer over a 1H frequency range of 5 kHz and a 15N frequency width of 25.4 kHz centered at δ = −200 ppm relative to CH315NO2 as external reference. 16 transients were acquired into 1024 data points for each of 128 t1 increments. The total acquisition time was 1 h for each 2D [1H, 15N] HSQC data set. 1H NMR spectra for dynamic solution experiments were acquired on a 400 MHz Varian spectrometer at 298 K. An equimolar solution of ligand 9 and [Cu(I)-9]PF6 was prepared in CD3OD at a total ligand concentration of 15.0 mM. The stock solution was diluted to final concentrations of 10.8, 7.2 and 3.6 mM of ligand 9. Full line shape analysis was performed with the gNMR software package22 using the spectra of the uncomplexed ligand 9 and [Cu(I)-9]PF6 as reference.
X-ray structural determination
X-ray quality crystals of [Cu(I)-9]PF6 were obtained from recrystallization in MeOH. Crystals were coated with ParatoneN oil, suspended in a small fiber loop, and placed in a cooled nitrogen gas stream at 173 K on a Bruker D8 APEX II CCD sealed tube diffractometer with graphite monochromated Mo-Kα (0.71073 Å) radiation. Data collection, indexing, and initial cell refinements were carried out using APEX II software.28 Frame integration and final cell refinements were done using the SAINT software.29 All structures were solved using direct methods and difference Fourier techniques (SHELXTL, V6.12).30Hydrogen atoms were placed in their expected chemical positions and were included in the final cycles of least-squares with isotropic Uij's related to the atom's ridden upon. All non-hydrogen atoms were refined anisotropically except for the disordered F atoms of the PF6 anion. Additional details of data collection and structure refinement are given in Tables S3 and S4.† The ORTEP drawing was generated with PLATON crystallographic software.31 Supplementary crystallographic data (CCDC 806655) can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request.cif.
Acknowledgements
Financial support from the National Institutes of Health (R01GM067169) is gratefully acknowledged. We thank Dr. Leslie T. Gelbaum for the HSQC NMR data and M. Thomas Morgan for suggesting a simplified synthetic protocol for the conversion of intermediate 6 to 7.
Notes and references
- J. Cody and C. J. Fahrni, Tetrahedron, 2004, 60, 11099 CrossRef CAS; R. F. H. Viguier and A. N. Hulme, J. Am. Chem. Soc., 2006, 128, 11370 CrossRef CAS; M. Taki, S. Iyoshi, A. Ojida, I. Hamachi and Y. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5938 CrossRef CAS.
- L. C. Yang, R. McRae, M. M. Henary, R. Patel, B. Lai, S. Vogt and C. J. Fahrni, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11179 CrossRef CAS; L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS; M. Verma, A. F. Chaudhry, M. T. Morgan and C. J. Fahrni, Org. Biomol. Chem., 2010, 8, 363 RSC.
- J. L. Bricks, A. Kovalchuk, C. Trieflinger, M. Nofz, M. Büschel, A. I. Tolmachev, J. Daub and K. Rurack, J. Am. Chem. Soc., 2005, 127, 13522 CrossRef CAS; M. Zhang, Y. Gao, M. Li, M. Yu, F. Li, L. Li, M. Zhu, J. Zhang, T. Yi and C. Huang, Tetrahedron Lett., 2007, 48, 3709 CrossRef CAS; W. Y. Lin, L. L. Long, L. Yuan, Z. M. Cao and J. B. Feng, Anal. Chim. Acta, 2009, 634, 262 CrossRef CAS; A. J. Weerasinghe, C. Schmiesing, S. Varaganti, G. Ramakrishna and E. Sinn, J. Phys. Chem. B, 2010, 114, 9413 CrossRef CAS.
- K. Rurack, M. Kollmannsberger, U. Resch-Genger and J. Daub, J. Am. Chem. Soc., 2000, 122, 968 CrossRef CAS; K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116 RSC.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651 CrossRef CAS.
- A. F. Chaudhry, M. Verma, M. T. Morgan, M. M. Henary, N. Siegel, J. M. Hales, J. W. Perry and C. J. Fahrni, J. Am. Chem. Soc., 2010, 132, 737 CrossRef CAS.
- A. P. De Silva, S. A. De Silva, A. S. Dissanayake and K. R. A. S. Sandanayake, Chem. Commun., 1989, 1054 RSC; A. P. De Silva and H. Q. Nimal Gunaratne, Chem. Commun., 1990, 186 RSC; A. P. De Silva, H. Q. N. Gunaratne and G. E. M. Maguire, Chem. Commun., 1994, 1213 RSC; A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson and M. Nieuwenhuizen, Chem. Commun., 1996, 1967 RSC; K. Rurack, J. L. Bricks, B. Schulz, M. Maus, G. Reck and U. Resch-Genger, J. Phys. Chem. A, 2000, 104, 6171 CrossRef CAS; K. Rurack, U. Resch-Genger, J. L. Bricks and M. Spieles, Chem. Commun., 2000, 2103 RSC.
- C. J. Fahrni, L. C. Yang and D. G. VanDerveer, J. Am. Chem. Soc., 2003, 125, 3799 CrossRef CAS.
- A. W. Addison, Inorg. Chim. Acta, 1989, 162, 217 CrossRef CAS.
- J. Cody, S. Mandal, L. C. Yang and C. J. Fahrni, J. Am. Chem. Soc., 2008, 130, 13023 CrossRef CAS.
- M. Verma, A. F. Chaudhry and C. J. Fahrni, Org. Biomol. Chem., 2009, 7, 1536 RSC.
- M. A. Clark, K. Duffy, J. Tilbrewala and S. J. Lippard, Org. Lett., 2003, 5, 2051 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CAS.
- D. A. Deranleau, J. Am. Chem. Soc., 1969, 91, 4044 CrossRef CAS.
- M. M. Bernardo, R. R. Schroeder and D. B. Rorabacher, Inorg. Chem., 1991, 30, 1241 CrossRef CAS.
- M. R. Malachowski, M. Adams, N. Elia, A. L. Rheingold and R. S. Kelly, J. Chem. Soc., Dalton Trans., 1999, 2177 RSC; K. K. Nanda, A. W. Addison, R. J. Butcher, M. R. McDevitt, T. N. Rao and E. Sinn, Inorg. Chem., 1997, 36, 134 CrossRef CAS; M. J. Schilstra, P. J. Birker, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1982, 21, 2637 CrossRef CAS.
- G. Binsch, J. B. Lambert, B. W. Roberts and J. D. Roberts, J. Am. Chem. Soc., 1964, 86, 5564 CrossRef CAS.
- A. Bax, R. H. Griffey and B. L. Hawkins, J. Magn. Reson., 1983, 55, 301 CAS.
- E. Shchori, J. Jagur-Grodzinski, Z. Luz and M. Shporer, J. Am. Chem. Soc., 1971, 93, 7133 CrossRef CAS; J. L. Dye, C. W. Andrews and J. M. Ceraso, J. Phys. Chem., 1975, 79, 3076 CrossRef CAS; E. Schmidt and A. I. Popov, J. Am. Chem. Soc., 1983, 105, 1873 CrossRef CAS.
- A. Delville, H. D. H. Stoever and C. Detellier, J. Am. Chem. Soc., 1985, 107, 4172 CrossRef CAS.
-
P. H. M. Budzelaar, gNMR Version 5.0, Adept Scientific PLC, Letchworth, UK, 2002. This software calculates the full bandshape of signals that are broadened as a result of chemical exchange Search PubMed.
- M. Pons and O. Millet, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 38, 267 CrossRef CAS.
- P. Maslak and W. H. Chapman, J. Org. Chem., 1996, 61 Search PubMed; G. W. Dombrowski, J. P. Dinnocenzo, P. A. Zielinski, S. Farid, Z. M. Wosinska and I. R. Gould, J. Org. Chem., 2005, 70, 3791 CrossRef CAS.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440 CrossRef CAS; R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CAS.
- J. Enderlein and R. Erdmann, Opt. Commun., 1997, 134, 371 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461 CrossRef.
-
APEX II, Bruker AXS, Inc., Madison, WI, 2005 Search PubMed.
-
SAINT v. 6.45A, Bruker AXS, Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112.
-
A. Platon, Multipurpose Crystallographic Tool, A. L. Spek, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.