DOI:
10.1039/C0SC00607F
(Edge Article)
Chem. Sci., 2011,
2, 1141-1144
Spiro-2,2′-bichroman-based bisoxazoline (SPANbox) ligands for ZnII-catalyzed enantioselective hydroxylation of β-keto esters and 1,3-diester†
Received
3rd December 2010
, Accepted 11th March 2011
First published on 30th March 2011
Abstract
A new type of spiro bisoxazoline ligand (SPANbox) has been developed on the basis of the spiro-2,2′-bichroman motif and bisoxazoline chelating units. The Zn(II) complexes of SPANbox showed excellent activity (98–99% yield) and enantioselectivity (90–98% ee) in the catalysis of hydroxylation of various β-keto esters and 1,3-diester with catalyst loading of 0.1–5 mol%. The performance of SPANbox in the present catalytic system was found to be significantly superior to the commonly used classic bisoxazoline ligands, such as (R,R)-Ph-Box or (S,S)-tBu-PyBox, indicating the advantage of the present spiro ligand skeleton.
Introduction
Enantioenriched α-hydroxyl-β-keto ester and 2-hydroxy-1,3-diester are key intermediates for the synthesis of various biologically important molecules.1,2(S)-5-Chloro-2-hydroxy-1-oxo-2,3-dihydro-1H-indene-2-carboxylic acid ester has been used as the key intermediate for the synthesis of chiral agrochemical Indoxacarb.3,4e The catalytic enantioselective hydroxylation of β-keto ester and 1,3-diester represents one of the most direct approach for their synthesis.4 Although several excellent catalytic systems including using chiral Lewis acids or organocatalysts have been reported,4 it seems that the activity of the catalysis still remains a challenging issue since high loading of catalyst and long reaction time have to be applied in order to realize high yield and enantioselectivity of the hydroxylation. In the context of chiral ligand design, spiro backbone has been recognized as one of the privileged structures for the construction of chiral ligands.5 On the other hand, bisoxazoline ligands (Box) represent one type of the most versatile chiral inducers in various transition metal-catalyzed reactions.6 As our ongoing endeavor to seek novel spiro chiral ligands,7 herein we communicate our preliminary results on the development of one type of chiral bisoxazoline ligands (SPANbox,1) with spiro-2,2′-bichroman motif8 by taking the advantages of spiro backbone of SPANphos9 and chelating units of Box,6 and their excellent performance in ZnII-catalyzed enantioselective hydroxylation of β-keto esters and 1,3-diester with racemic oxaziridine as the oxidant (Chart 1).
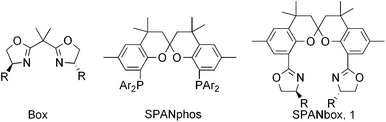 |
| | Chart 1 Working hypothesis for ligand design: from Box to SPANbox 1. | |
Results and discussion
Synthesis of spiro-bisoxazoline ligands
As shown in Scheme 1, the synthesis of enantiopure SPANbox ligands 1a–h is quite simple and straightforward by a four-step reaction sequence from racemic 4,4,4′,4′,6,6′-hexamethylspiro- 2,2′-bichroman (2) readily obtained in large scale by acid-catalyzed reaction of p-cresol with acetone.10 The bromination of 2 with NBS in DMF at r.t. afforded its dibromo derivative 3 in 96% yield. The dibromide 3 was further transformed to dicarboxylic acid 4 in 98% yield by reacting with nBuLi first and then CO2 at −78 °C. The condensation of 4 with a variety of enantiopure (S)-amino alcohols gave the corresponding hydroxy amides 5a–h in moderate to high yields. The two diastereomers of (S,R,S)-5a–h and (S,S,S)-5a–h could be readily separated by flash chromatography. The resulting enantiopure hydroxy amides (S,R,S)-5a–h and (S,S,S)-5a–g were finally converted into the corresponding bisoxazolines 1a–h by treatment with different methods according to the structural variation of 5 (for details, see ESI†).11 The absolute configuration of the spiro atom in (−)-1a was determined by X-ray diffraction analysis to be R (see ESI†).
![Synthesis of SPANbox [(S,S,S)-1 and (S,R,S)-1].](/image/article/2011/SC/c0sc00607f/c0sc00607f-s1.gif) |
| | Scheme 1 Synthesis of SPANbox [(S,S,S)-1 and (S,R,S)-1]. | |
Optimization of reaction conditions
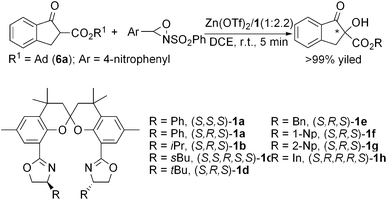
With the spiro bisoxazoline ligands 1a–h in hand, we then investigated their asymmetric induction in Lewis acid-catalyzed hydroxylation by using benzo-β-keto 1-admantyl ester 6a as a model substrate with a catalyst loading of 5 mol%. After screening of a variety of reaction conditions (see ESI, Tables S1–S5†) including the metal precursor, metal/ligand ratio, oxidant, solvent, and temperature, the reaction performed with Zn(OTf)2/(S,R,S)-1a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2) in 1,2-dichloroethane (DCE) at room temperature with racemic 3-(4-nitrophenyl)-2-(phenylsulfonyl)-1,2-oxaziridine as the oxidant turned out to be optimal. The complete conversion of substrate was realized within 5 min. The chirality at the spiro backbone of 1 was found to have a significant impact on the asymmetric induction of the catalysis (Table 1, entry 1 vs. 2). The combination of an R configuration of the spiro backbone and S,S configurations of the oxazoline moieties was disclosed as a matched case (entry 2). Further examination of the substituent effect of the oxazoline moieties in (S,R,S)-1a–h (entries 2–9) on the catalysis revealed that the ZnII complex of ligand (S,R,S)-1a having phenyl groups in the oxazoline moieties was the best in terms of both reactivity and enantioselectivity, affording the corresponding hydroxylation product (R)-7a in >99% yield with up to 99% ee (entry 2). Decrease of the catalyst loading to 1 mol% did not obviously deteriorate the enantioselectivity and the yield of the reaction (entry 10). It is noteworthy that the application of widely used bisoxazoline ligands5 such as (R,R)-Ph-Box or (S,S)-tBu-PyBox in the catalysis only afforded poor enantioselectivity (4–36% ee) although the activity is excellent (entries 11–13), indicating the advantage of the present ligand skeleton in the catalytic system.
:2.2) in 1,2-dichloroethane (DCE) at room temperature with racemic 3-(4-nitrophenyl)-2-(phenylsulfonyl)-1,2-oxaziridine as the oxidant turned out to be optimal. The complete conversion of substrate was realized within 5 min. The chirality at the spiro backbone of 1 was found to have a significant impact on the asymmetric induction of the catalysis (Table 1, entry 1 vs. 2). The combination of an R configuration of the spiro backbone and S,S configurations of the oxazoline moieties was disclosed as a matched case (entry 2). Further examination of the substituent effect of the oxazoline moieties in (S,R,S)-1a–h (entries 2–9) on the catalysis revealed that the ZnII complex of ligand (S,R,S)-1a having phenyl groups in the oxazoline moieties was the best in terms of both reactivity and enantioselectivity, affording the corresponding hydroxylation product (R)-7a in >99% yield with up to 99% ee (entry 2). Decrease of the catalyst loading to 1 mol% did not obviously deteriorate the enantioselectivity and the yield of the reaction (entry 10). It is noteworthy that the application of widely used bisoxazoline ligands5 such as (R,R)-Ph-Box or (S,S)-tBu-PyBox in the catalysis only afforded poor enantioselectivity (4–36% ee) although the activity is excellent (entries 11–13), indicating the advantage of the present ligand skeleton in the catalytic system.
Table 1 Optimization of SPANbox ligands for ZnII-catalyzed enantioselective hydroxylation of benzo-β-keto ester 6aa
|
|
| Entry |
Ligand, 1 |
s/cb |
ee [%]c |
|
Conditions: [Zn(OTf)2] = 12.5 mM; [1]= 27.5 mM (ZnII/1 = 1/2.2); [6a] = 0.25 M.
Substrate/catalyst molar ratio.
Determined by chiral HPLC. The absolute configuration in parenthesis was determined by comparison of [α]D with that of known data.
1-Np = naphthalen-1-ylmethyl.
2-Np = naphthalen-2-ylmethyl.
In = (1S,2R)-indan-1,2-yl.
|
| 1 |
(S,S,S)-1a |
20 |
26 (R) |
| 2 |
(S,R,S)-1a |
20 |
99 (S) |
| 3 |
(S,R,S)-1b |
20 |
86 (S) |
| 4 |
(S,S,R,S,S)-1c |
20 |
93 (S) |
| 5 |
(S,R,S)-1d |
20 |
51 (S) |
| 6 |
(S,R,S)-1e |
20 |
21 (S) |
| 7 |
(S,R,S)-1fd |
20 |
7 (S) |
| 8 |
(S,R,S)-1ge |
20 |
8 (S) |
| 9 |
(S,R,R,R,S)-1hf |
20 |
26 (S) |
| 10 |
(S,R,S)-1a |
100 |
97 (S) |
| 11 |
(R, R)-Ph-Box |
20 |
4 (S) |
| 12 |
(S,S)-tBu-PyBox
|
20 |
16 (R) |
| 13 |
(S,S)-Ph-PyBox
|
20 |
36 (R) |
The scope of substrates for catalytic enantioselective hydroxylation
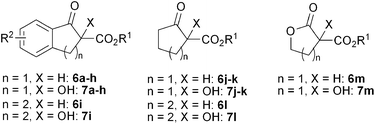
With the optimized catalyst ZnII/(S,R,S)-1a in hand, the hydroxylation of a variety of β-keto esters including five- and six-membered cyclic derivatives were subsequently carried out with 0.1–5 mol% catalyst loading at room temperature. As shown in Table 2, the substrates bearing a more sterically bulky tert-butyl or 1-admantyl group afforded the products in much higher enantioselectivities than the methyl derivative, indicating the critical importance of steric hindrance of ester groups (entries 1–3). It is obvious that the electronic properties of the substituent at a phenyl ring of benzo-β-keto esters (6a, 6d–h) do have some extent of impact on the enantioselectivity of the catalysis (entries 4–8 vs. 1) with the electron-donating substituent being favorable for the enantioselectivity. Product 7h, an homologue of the key intermediate for the synthesis of chiral agrochemical Indoxacarb,3 was obtained by the reaction of substrate 6h in 98% yield with 96% ee (entry 8), representing one of the best results reported so far for this important molecule.4 It should be noted that when the catalyst loading of Zn(OTf)2/(S,R,S)-1a was further reduced to 0.5–0.1 mol%, the hydroxylation of 6a still proceeded efficiently, affording (R)-7a in >99% yield with ee values of 95% and 90%, respectively (entry 9–10). The reaction of benzo-β-keto ester with six-membered ring (6i) is a little bit sluggish, affording the corresponding hydroxyl ester 7i with 94% ee in >99% yield in the presence of 5 mol% ZnII/(S,R,S)-1a (entry 11). The reactions of non-aryl substituted cyclic β-keto esters (6j–l) with either five- or six-membered rings also proceeded with high enantioselectivity albeit with relatively lower activity of 6l (entries 12–14). Similarly, the hydroxylation of cyclic 1,3-diester (6m) yielded the corresponding 2-hydroxy-1,3-diester 7m in excellent yield and enantioselectivity (99% ee) (entry 15 in Table 2).
|
|
| Entry |
6
|
n |
R1 |
R2 |
s/cb |
Time [min] |
Yield [%]c |
ee [%]d |
|
See conditions shown in the footnote of Table 1.
Substrate/catalyst molar ratio.
The yield of isolated product.
Determined by chiral HPLC or GC. The absolute configuration determined by comparison of the [α]D with those of reported data.
The absolute configuration not established.
|
| 1 |
6a
|
1 |
Ad
|
H |
100 |
5 |
>99 |
97(S)
|
| 2 |
6b
|
1 |
t
Bu
|
H |
100 |
5 |
>99 |
94(S)
|
| 3 |
6c
|
1 |
Me |
H |
100 |
5 |
>99 |
90(S)
|
| 4 |
6d
|
1 |
Ad
|
5-F
|
100 |
5 |
>99 |
93e |
| 5 |
6e
|
1 |
Ad
|
5-Cl
|
100 |
5 |
>99 |
95e |
| 6 |
6f
|
1 |
Ad
|
5-Me
|
100 |
5 |
>99 |
97e |
| 7 |
6g
|
1 |
Ad
|
5-MeO
|
100 |
5 |
>99 |
96e |
| 8 |
6h
|
1 |
Ad
|
4-Cl
|
20 |
5 |
98 |
96e |
| 9 |
6a
|
1 |
Ad
|
H |
200 |
10 |
>99 |
95(S)
|
| 10 |
6a
|
1 |
Ad
|
H |
1000 |
45 |
>99 |
90(S)
|
| 11 |
6i
|
2 |
Ad
|
— |
20 |
45 |
>99 |
94(S)
|
| 12 |
6j
|
1 |
Ad
|
— |
100 |
5 |
>99 |
97e |
| 13 |
6k
|
1 |
t
Bu
|
— |
100 |
5 |
>99 |
95(S)
|
| 14 |
6l
|
2 |
Ad
|
— |
20 |
180 |
98 |
93e |
| 15 |
6m
|
1 |
Ad
|
— |
100 |
300 |
>99 |
99e |
Mechanistic consideration
To gain an insight into the mechanistic aspect of the catalysis, the nonlinear effect12 in the catalytic system was investigated at room temperature by examining the relationship between the enantiopurity of the ligand used in corresponding product (S)-7a. On the other hand, comparison of 1H NMR spectra (see ESI†) of ligand (S,R,S)-1a and its Zn(OTf)2 complex prepared in situ in CDCl3, as well as the MS data (see ESI†), clearly shows the formation of a 1:1 complex of (S,R,S)-1a with Zn(OTf)2 albeit in the presence of excessive (S,R,S)-1a. These data are well consistent with an indication that the 1:1 complex of ZnII with SPANbox might be a catalytically active species, albeit 1:2.2 ZnII-ligand ratio is optimal for catalysis. Accordingly, a plausible asymmetric induction model can be tentatively proposed (Fig. 1b) on the basis of the molecular structure of (S,R,S)-1a (see ESI†) and absolute configuration of the corresponding resultant product 7a.
![(a) Plot of ee values of 7aversus ee values of (S,R,S)-1a with [6a] = 0.25 M, [ZnII] = 0.0125 M and ZnII/(R,S,R)-1a = 1 : 2.2 at 25 °C. (b) Proposed asymmetric induction model.](/image/article/2011/SC/c0sc00607f/c0sc00607f-f1.gif) |
| | Fig. 1 (a) Plot of ee values of 7aversus ee values of (S,R,S)-1a with [6a] = 0.25 M, [ZnII] = 0.0125 M and ZnII/(R,S,R)-1a = 1:2.2 at 25 °C. (b) Proposed asymmetric induction model. | |
Conclusions
In summary, a new class of bisoxazoline ligand (SPANbox, 1) based on the spiro-2,2′-bichroman backbone has been developed by simple transformations of readily available starting materials. Its cationic ZnII complex was found to be highly efficient in the catalysis of hydroxylation of a broad range of β-keto esters and 1,3-diester, and the corresponding optically active hydroxyl products with quaternary carbon centers were obtained with ee values of up to 99%. The excellent performance of this type of ligand in catalysis will stimulate future efforts to explore the new applications of these ligands in other transition metal-catalyzed asymmetric reactions.
Experimental
The reaction of 6a was taken as a typical example for the procedure of enantioselective hydroxylation. Zn(OTf)2 (0.9 mg, 0.0025 mmol) and (S,R,S)-1a (3.4 mg, 0.0055 mmol) were weighed in a nitrogen-filled glovebox and transferred into a pre-dried Schlenk tube equipped with a Teflon-coated magnetic stirring bar. The Schlenk tube was further vacuumized for 2 h before dry 1,2-dichloroethane (1.0 mL) was added under argon atmosphere. The resultant solution was stirred for 1 h and the substrate β-keto ester 6a (77.6 mg, 0.25 mmol) was added to the solution. After stirring for an additional 30 min, the oxidant 3-(4-nitrophenyl)-2-(phenylsulfonyl)-1,2-oxaziridine (91.9 mg, 0.30 mmol) was added to the solution in one portion. The mixture was stirred at room temperature for 5 min. The reaction was quenched by addition of saturated aqueous NaHSO3 solution. The mixture was then extracted with dichloromethane, the combined organic phase was washed with brine and then dried over Na2SO4. After removal of the solvent under vacuum evaporation, the residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate as the eluent to afford 7a as a colorless oil (97.0 mg, yield >99%). Colorless oil, 98% ee; [α]20D = +19.7 (c = 0.89, CHCl3) [lit.4d [α]20D = +19.7 (c = 0.60, CHCl3) for 91% ee (S)]; 1H NMR (300 MHz, CDCl3) δ 7.79 (1H, d, J = 7.8 Hz), 7.67–7.62 (1H, m), 7.48–7.38 (2H, m), 4.03 (1H, br), 3.65 (1H, d, J = 17.4 Hz), 3.22 (1H, d, J = 17.1 Hz), 2.11 (3H, s), 1.97–1.95 (6H, m), 1.60–1.59 (6H, m); 13C NMR (100 MHz, CDCl3) δ 201.4, 170.1, 152.3, 135.7, 133.9, 127.8, 126.2, 125.0, 83.8, 80.5, 40.8, 39.5, 35.8, 30.7; IR (neat) ν 3476, 2912, 2852, 1740, 1608, 1457, 1249, 1196, 1103, 1049, 964, 881, 754, 695, 669 cm−1; EI-MS, m/z = 243, 195, 179, 152, 135, 103, 95, 76, 51. The enantiomeric excess was determined by HPLC on Chiralpak AD-H column, hexane: isopropanol = 90: 10, flow rate: 1.0 mL min−1, 254 nm, tR1 = 15.6 min (major), tR2 = 24.6 min (minor).
Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (grant nos. 20821002, 20923005, 20972176), the Chinese Academy of Sciences, the Major Basic Research Development Program of China (grant no. 2010CB833300), the Science and Technology Commission of Shanghai Municipality are gratefully acknowledged.
Notes and references
- J. Christoffers, A. Baro and T. Werner, Adv. Synth. Catal., 2004, 346, 143 CrossRef CAS.
-
G. M. Coppola, H. F. Schuster, α- Hydroxyacids in Enantioselective Synthesis, VCH, Weinheim, 1997 Search PubMed.
-
G. D. Annis, S. F. McCann, R. Shapiro, WO 95/29171, PCT/US95/04321, 1995.
- For reviews on catalytic enantioselective α-heterofunctionalization of carbonyl compounds, see:
(a) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292 CrossRef CAS;
(b) G. Guillena and D. J. Ramón, Tetrahedron: Asymmetry, 2006, 17, 1465 CrossRef CAS;
(c) T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917 Search PubMed;
(d) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637 CrossRef CAS . For recent examples, see;
(e) M. R. Acocella, O. G. Mancheno, M. Bella and K. A. Jørgensen, J. Org. Chem., 2004, 69, 8165 CrossRef CAS;
(f) P. Y. Toullec, C. Bonaccorsi, A. Mezzetti and A. Togni, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5810 CrossRef CAS;
(g) T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488 CrossRef;
(h) D. S. Reddy, N. Shibata, J. Nagai, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2009, 48, 803 CrossRef;
(i) M. Lu, D. Zhu, Y. Lu, X. Zeng, B. Tan, Z. Xu and G. Zhong, J. Am. Chem. Soc., 2009, 131, 4562 CrossRef CAS;
(j) A. M. R. Smith, D. Billen and K. K. Hii, Chem. Commun., 2009, 3925 RSC;
(k) A. M. R. Smith, H. S. Rzepa, A. J. P. White, D. Billen and K. K. Hii, J. Org. Chem., 2010, 75, 3085 CrossRef CAS;
(l) B. Gong, Q. Meng, T. Su, M. Lian, Q. Wang and Z. Gao, Synlett, 2009, 2659 CAS;
(m)
A. L. Casalnuovo, WO 03/002255, PCT/US02/17755, 2003;
(n) J. J. Jiang, J. Huang, D. Wang, M. X. Zhao, F. J. Wang and M. Shi, Tetrahedron: Asymmetry, 2010, 21, 794 CrossRef CAS;
(o) S. H. Cao and M. Shi, Tetrahedron: Asymmetry, 2010, 21, 2675 CrossRef CAS;
(p) M. M. Lian, Z. Li, J. Du, Q. W. Meng and Z. X. Gao, Eur. J. Org. Chem., 2010, 6525 CrossRef CAS.
- For leading references, see:
(a) A. S. C. Chan, W. H. Hu, C. C. Pai, C. P. Lau, Y. Z. Jiang, A. Q. Mi, M. Yan, J. Sun, R. L. Lou and J. G. Deng, J. Am. Chem. Soc., 1997, 119, 9570 CrossRef CAS;
(b) M. A. Arai, M. Kuraishi, T. Arai and H. Sasai, J. Am. Chem. Soc., 2001, 123, 2907 CrossRef CAS;
(c) T. Tsujihara, K. Takenaka, K. Onitsuka, M. Hatanaka and H. Sasai, J. Am. Chem. Soc., 2009, 131, 3452 CrossRef CAS;
(d) S.-F. Zhu, Y. Cai, H. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS;
(e) T. Kato, K. Marubayashi, S. Takizawa and H. Sasai, Tetrahedron: Asymmetry, 2004, 15, 3693 CrossRef CAS;
(f) J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS;
(g) J.-B. Xie, J.-H. Xie, X.-Y. Liu, Q.-Q. Zhang and Q.-L. Zhou, Chem.–Asian J., 2011, 6, 899 CrossRef CAS . For reviews, see;
(h) J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS;
(i) G. B. Bajracharya, M. A. Arai, P. S. Koranne, T. Suzuki, S. Takizawa and H. Sasai, Bull. Chem. Soc. Jpn., 2009, 82, 285 CrossRef CAS;
(j) K. Ding, Z. Han and Z. Wang, Chem.–Asian J., 2009, 4, 32 CrossRef CAS.
-
(a) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS;
(b) E. J. Corey, N. Imai and H. Zhang, J. Am. Chem. Soc., 1991, 113, 728 CrossRef CAS . For reviews, see;
(c) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS;
(d)
A. Pfaltz, in Asymmetric Synthesis-The Essentials, ed. M. Christmann and S. Braese, Wiley-VCH, Weinheim, 2nd edn, 2008, pp. 139–143 Search PubMed.
-
(a) Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345 CrossRef CAS;
(b) Y. Zhang, Z. Han, F. Li, K. Ding and A. Zhang, Chem. Commun., 2010, 46, 156 RSC.
- P. T. Mora and T. Szeki, J. Am. Chem. Soc., 1950, 72, 3009 CrossRef CAS.
- For leading work in this area, see:
(a) Z. Freixa, M. S. Beentjes, G. D. Batema, C. B. Dieleman, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, J. Fraanje, K. Goubitz and P. W. N. N. van Leeuwen, Angew. Chem., Int. Ed., 2003, 42, 1284 CrossRef CAS;
(b) Z. Freixa, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2005, 44, 4385 CrossRef CAS;
(c) X. Sala, E. J. Garcia Suarez, Z. Freixa, J. Benet-Buchholz and P. W. N. M. van Leeuwen, Eur. J. Org. Chem., 2008, 6197 CrossRef CAS.
-
(a) A. Ryabov, Neftekhimiya, 1987, 27, 21;
(b) A. J. Caruso and J. L. Lee, J. Org. Chem., 1997, 62, 1058 CrossRef . See also ref. 8.
- For method A (5a and 5d), see:
(a) U. Iserloh, D. P. Curran and S. Kanemasa, Tetrahedron: Asymmetry, 1999, 10, 2417 CrossRef CAS for method B (5b–c and 5e–g), see;
(b) B. Method, M. Ogasawara, K. Yoshida, H. Kamei, K. Kato, Y. Uozumi and T. Hayashi, Tetrahedron: Asymmetry, 1998, 9, 1779 CrossRef CAS for method C (5h), see;
(c) M. Kurosu, J. R. Porter and M. A. Foley, Tetrahedron Lett., 2004, 45, 145 CrossRef CAS.
-
(a) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef;
(b) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthesis and characterisation of all compounds. CCDC reference number 787243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00607f |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.