DOI:
10.1039/C0SC00592D
(Edge Article)
Chem. Sci., 2011,
2, 752-759
Controlling dynamic stereoisomerism in transition-metal folded baskets†
Received
25th November 2010
, Accepted 9th January 2011
First published on 28th January 2011
Abstract
We synthesized chiral molecular baskets (R3/S3)-1, containing three pyridine rings at the rim of a bowl-shaped platform. Each pyridine was tethered to the platform via a CH(CH)3 stereogenic center. In line with prior studies, we found that chiral basket (R3)-1 would coordinate to Ag(I) cation forming a Ag(I)–(R3)-1 complex (ΔG° = −5.9 kcal mol−1 at 298.0 K). Furthermore, we used spectroscopic methods (1H NMR, UV-Vis and CD) to establish that the CH(CH)3 stereogenic center with R configuration in Ag(I)–(R3)-1 would direct the twisting of the pyridine rings at the rim in a clockwise orientation (P configuration). UV-Vis and CD spectra of Ag(I)–(R3)-1(P) were also computed (TD-DFT; BHLYP/SV(P),TZVP), showing good agreement with the experimental data. Controlling dynamic stereoisomerism in basket-like systems is important and the results of this study will be useful for examining the relationship between chiral recognition and reactivity in gated and confined environments.
Introduction
There has been significant interest in developing methods for controlling the axial/propeller type of chirality within various organic1–4 and metal-mediated coordination systems.5–11 Hence, placing an electrophilic metal in a chiral/dynamic environment affords a greater discrimination of substrates12 and is essential for developing chiral discrimination technologies17–20 and expanding the scope of stereoselective synthesis.21–24 In particular, transition-metal complexes of multidentate nitrogen-containing ligands (i.e. tris(pyridylmethyl)amine (TPA), Fig. 1A) have been scrutinized for understanding the activation of C–H bonds25–27 and obtaining stimuli-responsive switches/sensors.19,28,29 In TPA coordination systems, the propeller-like chirality was controlled with an alkyl substituent: the alkyl group would, as a relay auxiliary,30 promote the formation of predominantly one twisted diastereomer (Fig. 1A).9 That is, the S stereogenic center was established9 to direct the formation of the M propeller-like stereogenic unit and vice versa (Fig. 1A). Transition-metal (Ag(I), Cu(I)) folded baskets32–35 are somewhat related to TPA ligands although they could also act as hosts. For our molecular baskets, three pyridine gates at the rim of the bowl-shaped platform coordinate to the electrophilic metal, adopting a propeller-like orientation (Fig. 1B). In Cu(I)-folded baskets, the fourth coordination site is occupied with a guest molecule protruding into the host's inner space and exchanging to/from it via a slippage mechanism.34 As a part of our goal toward using basket-like hosts for investigating encapsulation kinetics36,37 and their relation to chemical reactions occurring inside dynamic and confined environments, we were intrigued by the prospect of controlling stereoisomerism in this particular environment. Accordingly, we set to investigate the extent to which a stereogenic center at the “hinge” position directs the symmetric characteristics of the propeller-like stereogenic unit composed of three pyridine gates (Fig. 1B). Will the S or R methyl group at the “hinge” have a measurable effect on the M/P twisted orientation of the pyridines (Fig. 1B)? Our current study demonstrates that propagating stereochemical information inside Ag(I)-folded molecular baskets35 is indeed feasible: the pyridine groups in Ag(I)–(R)3-1 have been found to predominantly twist into a P type propeller, while an M propeller was observed for Ag(I)–(S)3-1.
 |
| | Fig. 1 (A) Chemical structure of α-MeTPA ligand and energy minimized structures of its Zn(II) complexes (MM, Spartan).9 (B) Chemical structure of (R3)-1 basket and DFT energy minimized structures (RI-BHLYP/SV(P),TZVP)13–16 of two conformational diastereomers Ag(I)–(R3)-1(M) and Ag(I)–(R3)-1(P). | |
Results and discussion
Synthesis
Chiral baskets (R3)-1/(S3)-1 and stereoisomeric model compounds (R)-2/(S)-2 were obtained following already published protocols31 for the asymmetric synthesis of enantiopure amines (R/S)-7 (Scheme 1). In accordance with such procedures, the delivery of a “hydride” reducing agent to the two diastereotopic faces of enantiopure N-tert-butanesulfinyl ketimine (RA)-5 occurs at a different rate, allowing the predominant formation of one diastereomer; note that we used subscript A for assigning the auxiliary sulfinyl unit. In particular, diisobutylaluminium hydride (DIBAL) was reacted with (RA)-5 to give compound (RA, R)-6 with excellent diastereoselectivity (d.s. > 99%, Scheme 1).31 Lithium tris-sec-butyl(hydrido)borate (L-selectride), however, reduced (RA)-5 to give epimeric (RS, S)-6 (d.s. > 95%, Scheme 1).31 Enantiopure amines (R/S)-7 were obtained upon the removal of the chiral auxiliary (CH3OH/6M HCl), and then each reacted with phthalic anhydride or tris-anhydride 8 to give stereoisomeric model compounds (R or S)-2 or chiral baskets (R3 or S3)-1, respectively.38
 |
| | Scheme 1 Synthetic routes for the preparation of chiral baskets (R3/S3)-1 and model compounds (R/S)-2; note that the preparation of (R/S)-7 was completed following already published procedures.31 | |
Computational studies of model compound (R)-2
Compound (R)-2 contains a pyridine ring tethered to the phthalimide unit via an R stereogenic CH(CH3) center (Fig. 2). In anticipation that this particular framework would mimic our baskets, we decided to examine the conformational dynamics as well as spectroscopic characteristics of (R/S)-2. The result of Monte Carlo conformational searches (MM3)39–41 suggested that two conformers A/B of (R)-2 should populate the equilibrium (Fig. 2).38 Notably, each conformer has its hinge C–H unit nearly eclipsed with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O phthalimide group and the plane of the pyridine ring, while the position of the pyridine nitrogen is either syn (A) or anti (B) with respect to the C–H. Solid-state structures of similar molecules have revealed the corresponding H–C–N–C(O) dihedral angle to be in the range of 4° to 19°.42 Additional density functional theory (DFT: RI-B3LYP/TZVP and RI-BHLYP/TZVP)13–16,43 gas phase calculations of (R)-2 were performed using TURBOMOLE 5.10.15 Conformer A was calculated to be the preferred orientation (0.7 kcal mol−1) by both levels of theory, corresponding to a 3
O phthalimide group and the plane of the pyridine ring, while the position of the pyridine nitrogen is either syn (A) or anti (B) with respect to the C–H. Solid-state structures of similar molecules have revealed the corresponding H–C–N–C(O) dihedral angle to be in the range of 4° to 19°.42 Additional density functional theory (DFT: RI-B3LYP/TZVP and RI-BHLYP/TZVP)13–16,43 gas phase calculations of (R)-2 were performed using TURBOMOLE 5.10.15 Conformer A was calculated to be the preferred orientation (0.7 kcal mol−1) by both levels of theory, corresponding to a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between conformations. Computing the Gibbs free energies (at 298 K) slightly reduces the energetic preference to 0.4 kcal mol−1 (RI-BHLYP). We further speculate that the C–H group is nearly eclipsed with the plane of the phthalimide ring for minimizing the unfavorable van der Waals type of strain between bulkier CH3 and CO groups44 (e.g. resembling axial (A1,3) strain).45
:1 ratio between conformations. Computing the Gibbs free energies (at 298 K) slightly reduces the energetic preference to 0.4 kcal mol−1 (RI-BHLYP). We further speculate that the C–H group is nearly eclipsed with the plane of the phthalimide ring for minimizing the unfavorable van der Waals type of strain between bulkier CH3 and CO groups44 (e.g. resembling axial (A1,3) strain).45
 |
| | Fig. 2 Chemical structure of (R)-2 model compound and the corresponding Newman projections with DFT energy minimized (RI-BHLYP/TZVP) structures of the two rotamers A and B of (R)-2. | |
1H NMR spectroscopic studies of model compound (R)-2
1H NMR spectrum of (R)-2 (400 MHz, 298.0 K) showed a single set of signals (Figure S7†); we used chemical shifts (δ, ppm) and the splitting characteristics of the nuclei to assign these signals (Fig. 2). Variable temperature 1H NMR measurements (298.0–193.0 K) revealed no decoalescence except for a slight broadening of resonances at lower temperatures (Figure S7†). Importantly, a 2D 1H-NMR NOESY spectrum of (R)-2 (0.16 M, CD3CN; 298.0 K) exhibited two prominent cross-signals corresponding to Ha and Hd protons in the vicinity of the CH3 group (Figure S13†).38 Indeed, the calculated distance between the pertinent nuclei is 2.1–2.4 Å (Fig. 2), consistent with the observed spin polarization transfers and providing some evidence for the rotamers A and B. The absence of decoalescence of 1H NMR signals at lower temperatures (Figure S7†), however, is consistent with A/B exchanging fast on the NMR time scale. In fact, there is a small calculated activation barrier for rotating the pyridine about the C–C single bond that is connecting this ring to the CH(CH3) group, thus supporting the notion about the fast conformational interconversion.
UV-Vis and CD spectroscopic studies of model compounds (R/S)-2
Model compounds (R/S)-2 encompass pyridine and phthalimide chromophores separated with a CH(CH3) group (Fig. 2). Five principal electronic transitions were reported to contribute to the UV-Vis spectrum of the phthalimide chromophore.46 In particular, transitions at 300 nm (ε = 1800 M−1 cm−1), 237 nm (ε = 12200 M−1 cm−1) and 221 nm (ε = 33200 M−1 cm−1), are of π → π* type and prominent; the last one has its transition electric dipole moment along the long axis of the molecule. The UV-Vis spectrum of pyridine as a chromophore,47–50 however, is similar to that of benzene and characterized with a broad band at ∼270 nm consisting of two almost degenerate electronic transitions: a symmetry allowed, but weak, π → π* transition at 260 nm (ε = 2000 M−1 cm−1) and a weaker n→ π* transition at longer wavelengths ∼270 nm; the corresponding electric transition moments are in plane for the π → π* state and perpendicular to the same plane for the n → π* state. The UV-Vis spectrum of compound (R)-2 (Fig. 3A, 298.0 K) contained three broad absorptions and a shoulder at approximately 295, 260, 225 and 240 nm. These bands could be qualitatively assigned to two chromophores, and for clarity, we labeled them as I, II, III and IV (Fig. 3A). Furthermore, we decided to compute the UV-Vis spectrum of the Boltzmann weighted (R)-2 using time-dependent density functional theory methods (TD-DFT, B3LYP/TZVP and BHLYP/TZVP, Fig. 3);13–16,38,43,51,52 The spectra were red-shifted (0.15 eV for B3LYP, 0.85 eV for B3LYP) and a 0.3 eV Gaussian line-broadening was applied to each excitation. The calculated spectra were normalized to the experimental peak at 220 nm. Our computational procedure for modeling these spectra mirrors previously successful work on systems of various sizes.54 Importantly, the electronic spectrum of (R)-2 (BHLYP) showed a fair agreement with the experimental data (Fig. 3A). After applying the appropriate shift, the four dominant electronic transitions contributing to the theoretical spectrum were found at 292 (I), 252 (II), 243 (III) and 220 (IV) nm. While these bands are in accordance with those reported in the literature (see above), electron density difference plots were still computed to confirm assignment (Fig. 3B).38 Electron density difference plots reveal the accumulation (green) and depletion (red) of the electron density for a particular transition by subtracting the electron density of the ground-state (S0) from the desired excited state.55
 |
| | Fig. 3 (A) UV-Vis (top) and CD (bottom) spectra of model compounds (R)-2 (0.029 mM) and (S)-2 (0.029 mM) in CH3CN at 298.0 K (black lines). Computed UV-Vis and CD spectra (TD-BHLYP/TZVP) of model compound (R)-2 (red line). (B) The electron density difference plots depicting I–IV electronic transitions.38 The contour values are ±0.001 au. | |
Circular dichroism (CD) spectroscopy provides information about the absolute configuration and conformation of molecules lacking a rotation–reflection (Sn) symmetry axis;56 in fact, the method is based on differential absorption of left and right circularly polarized light (AL ≠ AR).57 In the case at hand, the CD spectrum of (R/S)-2 revealed Cotton effects in the areas corresponding to the phthalimide (bands III and IV, Fig. 3) and pyridine (band II, Fig. 3) chromophores. While the excitonic coupling (EC-CD) of the two chromophores42,58,59 was equivocal, our computed CD spectrum (TD-BHLYP/TZVP) of the Boltzmann weighted (R)-1 was in decent agreement with the experimental one (Fig. 3A).38 Perhaps, accounting for explicit solvation60–62 of the conformers as well as their dynamics could give a more satisfactory theoretical spectrum though such tedious computational studies would be very time-consuming. Additionally, the only difference between the two conformations is the rotation of the pyridine moiety. Using DFT calculations (RI-BP86/TZVP),63,64 we found the energy barrier of rotation between the conformers to be only 2.2 kcal mol−1.38 The barrier is sufficiently low enough for rotation to occur at room temperature, and thus the CD calculations on our two static structures are likely insufficient in accurately describing the averaged CD spectra. We nonetheless decided to direct our efforts toward examining the transfer of chirality in Ag(I)-folded baskets using the TD-BHLYP/SV(P),TZVP level of theory.
Computational studies of chiral basket (R3)-1
Chiral molecular basket (R3)-1 contains three pyridine rings each tethered to the bowl-shaped platform via the CH(CH3) “hinge” group (Fig. 1B). In line with the conformational analysis of model (R3)-2 (Fig. 2), we reasoned that the hinge C–H bonds should adopt a nearly eclipsed position with the adjacent CO groups. Indeed, the sampling of the conformational space using Monte Carlo (MM3) protocol corroborated our prediction: four conformational isomers of (R3)-1 were identified,38 with the population bias somewhat inclined toward so-called (R3)-1three-in conformer (Fig. 4). The computed steric energies (Fig. 4) were, however, close enough to envisage a contribution from all four proposed conformers. Notwithstanding the complexity of the situation, we decided to computationally examine the Ag(I)–(R3)-1 basket having three pyridine rings restricted by dative bonds between Ag(I) and the pyridine nitrogens (Fig. 1B). DFT (RI-BHLYP/SV(P),TZVP) geometry optimizations of Ag(I)–(R3)-1 revealed two conformational diastereomers (Fig. 1B) based upon the orientation of the pyridine rings in either clockwise P or counter-clockwise M orientation. The P diastereomer of Ag(I)–(R3)-1 was computed to be considerably more stable (ΔE = 6.6 kcal mol−1) than the corresponding M diastereomer. That is to say, on the basis of the calculations, one should expect the relay of stereochemical information65,66 so that the R stereogenic center directs the exclusive formation of the P orientation and conversely, the S stereogenic center would favor M (Fig. 1B).
 |
| | Fig. 4 Energy minimized structures (MM3) of four conformers of basket (R3)-1, generated from a Monte Carlo conformational search.38 The presented steric energies (kcal mol−1) were computed for the gas phase and for water. | |
1H NMR spectroscopic studies of chiral baskets and their coordination to Ag(I) cation
1H NMR spectrum of basket (R3)-1 in CD3CN (400 MHz, 298.0 K) revealed a set of signals corresponding to a molecule with an averaged C3v symmetry (Fig. 5A); the assignment of the observed resonances was completed by considering chemical shift (δ, ppm) and coupling constants (J, Hz) of chemically/magnetically different nuclei (Fig. 5B). There was no decoalescence of the 1H NMR signals at low temperatures (298.0–193.0 K, Figure S18†). In accordance with the computed steric energies of the conformers of (R3)-1 (Fig. 4), we surmise that the activation barrier for their interconversion ought to be small. In addition, 2D 1H-NMR NOESY spectrum of (R3)-1 (400 MHz, 298.0 K) contained a distinct cross signal corresponding to Hb/CH3 protons in the vicinity, indicating the presence of a particular pyridine rotamer (see Figure S16†).38
 |
| | Fig. 5 (A) A series of 1H NMR spectra (400 MHz, 298.0 K) of (R3)-1 (3.40 mM) in CD3CN recorded upon incremental addition of (a) 0.0 (b) 0.1, (c) 0.2, (d) 0.3, (e) 0.4, (f) 0.5, (g) 0.6, (h) 0.7, (i) 0.8, (j) 0.9, (k) 1.0, (l) 1.1, (m) 1.2, (n) 1.3, (o) 1.4, (p) 1.5, (q) 1.8, (r) 2.1, (s) 2.4, (t) 2.7, and (u) 3.0 molar equivalents of Ag(BF4). (B) The nonlinear curve fitting (SigmaPlot) of the 1H NMR chemical shifts of Ha/b/c/d resonances to a 1:1 binding model.53 (C) Selected regions of 1H–1H NOESY spectrum (400 MHz, CD3CN, 200 ms mixing time) of a solution of Ag(I)–(R3)-1 at 298.0 K. | |
We recently reported studies32,35 describing the propensity of pyridine-containing baskets, akin to (R3/S3)-1, for coordinating to Ag(I) cation (ΔG° = −7.2 kcal mol−1 at 300.0 K). Accordingly, we titrated a stock solution of AgBF4 (169.5 mM) to (R3)-1 (3.40 mM) in CD3CN and followed the binding with 1H NMR spectroscopy (Fig. 5A). The signal for Ha shifted upfield (|Δδ| = 0.45 ppm) while the resonances for Hc/d protons shifted downfield (|Δδ| = 0.25–0.32 ppm) indicating the formation of the Ag(I)–(R3)-1 complex.35 Nonlinear least-squares analysis of the titration data53 suggested the stability constant Ka (298.0 K) to be 2.2 ± 0.7 × 104 (ΔG° = −5.9 kcal mol−1, Fig. 5B)); note that this stability constant Ka was obtained as an average from the analysis of four binding isotherms corresponding to Ha-d protons (Fig. 5B). Evidently, placement of the alkyl CH3 group at the hinge position did not have a substantial effect on the basket's propensity for coordinating to Ag(I). We then examined the conformational characteristics of Ag(I)–(R3)-1 with 2D NOESY (400 MHz, 298.0 K) spectroscopy. Two cross signals corresponding to protons Ha/He and Hd/CH3 were prominent (Fig. 5C), suggesting a close proximity of the corresponding nuclei (< 5 Å). Indeed, the more stable diastereomer of Ag(I)–(R3)-1 with the pyridine units twisted clockwise (P) was computed to contain Ha/He and Hd/CH3 protons within 2.55 Å and 2.30 Å, respectively (Fig. 6C).
UV-Vis and CD spectroscopic studies of chiral baskets and their coordination to Ag(I) cation
The electronic spectrum of (R3)-1 (Fig. 6A) was akin to that of model compound (R)-2 (Fig. 3A), with pronounced transitions at 290 (I), 260 (II), 245 (III) and 225 (IV) nm. In accordance with our prior discussion and literature precedent, bands I, III and IV can be formally ascribed to the phthalimide chromophore, while band II to the pyridine chromophore. The addition of stock solution of AgBF4 (0.13 mM) to (R3)-1 (0.013 mM) in CH3CN led to the formation of Ag(I)–(R3)-1, although the binding did not have a noticeable effect on the principal UV-Vis transitions (Fig. 6A). This, however, was not the case with the CD spectra in which the formation of Ag(I)–(R3)-1 was followed with the appearance of prominent Cotton effects at 260 (II), 245 (III) and 225 (IV) nm (Fig. 6A). In particular, there appeared a considerable increase in the intensity of the band II (at 260 nm), which is in line with the “fixation” of the pyridine chromophores following the complexation event. As expected, the coordination of basket (S3)-1 to Ag(I) cation gave rise to CD signals having completely opposite values (Fig. 6B) from those corresponding to Ag(I)–(R3)-1 (Fig. 6A), thereby validating the enantiomeric nature of the two stereoisomers. Furthermore, we computed UV-Vis and CD spectra of Ag(I)–(R3)-1 using time-dependent density functional theory (TD DFT, BHLYP/TZVP, Fig. 7A).38 Since the P conformation was determined (ΔE = 6.6 kcal mol−1, Fig. 1B) to entirely populate the equilibrium, there was no need to evaluate the spectroscopic characteristics of the M diastereomer (see Figure S27†). Notably, there is a good agreement between the computed and experimental spectra of Ag(I)–(R3)-1(P); there were four principal electronic transitions I–IV contributing to the theoretical UV-Vis spectrum (Fig. 7A). The corresponding Cotton effects were evaluated with the assistance of the electron density difference plots (Fig. 7B), corroborating our qualitative assignment of the bands: band II is for the most part associated with the pyridine rings, while bands III and IV are associated with the phthalimides. Evidently, the coordination of three pyridines in (R3)-1 to Ag(I) limited the conformational mobility of these chromophores, thereby affecting the overall intensity of band II. The phthalimide chromophores were also perturbed upon the formation of Ag(I)–(R3)-1(P) (Fig. 6A/7A). There, the increase in the intensity of the bands III and IV would imply the formation of an assembled and at the same time conformationally restricted system. Importantly, there is very good agreement between the computed and experimental CD spectra of Ag(I)–(R3)-1(P) and the experimental and computational data are consistent with the proposed chirality transfer and the formation of the energetically biased P diastereomer. At last, the computed CD spectrum of less stable Ag(I)–(R3)-1(M) stereoisomer (TD-BHLYP/SV(P),TZVP, Figure S27†) appeared as nearly a mirror image of the one corresponding to the P diastereomer (Fig. 7A) thereby corroborating the prominence of the propeller-like orientation of the pyridine chromophores and in line with the assigned sense of chirality.
 |
| | Fig. 6 (A) A series of UV-Vis (top) and CD (bottom) spectra (298.0 K) of (R3)-1 (0.013 mM) in CD3CN recorded upon incremental addition of (a) 0.0 (b) 0.1, (c) 0.2, (d) 0.3, (e) 0.4, (f) 0.5, (g) 0.6, (h) 0.7, (i) 0.8, and (j) 1.0, molar equivalents of Ag(BF4). (B) UV-Vis spectrum (298.0 K) of (S3)-1 (0.013 mM) (top) and a series of CD (bottom) spectra (298.0 K) of (S3)-1 (0.013 mM) in CD3CN recorded upon incremental addition of (a) 0.0 (b) 0.1, (c) 0.2, (d) 0.3, (e) 0.4, (f) 0.5, (g) 0.6, (h) 0.7, (i) 0.8, (j) 0.9, (k) 1.0 and (l) 1.1 molar equivalents of Ag(BF4); note that the addition of Ag(BF4) to (R3/S3)-1 caused enhancement in the intensity of II–IV bands (CD). | |
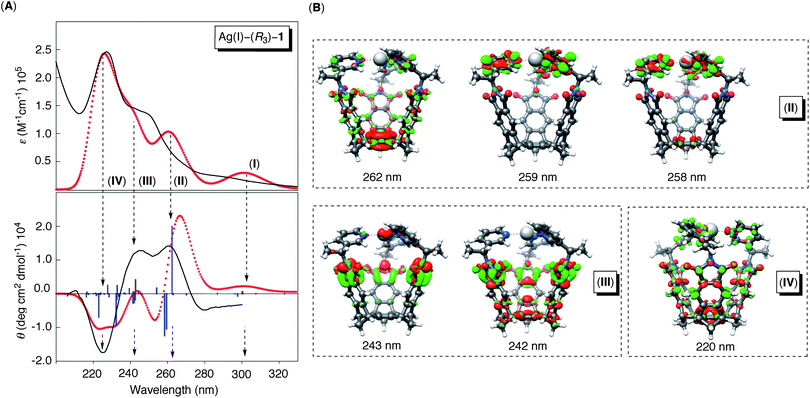 |
| | Fig. 7 (A) Computed (TD-BHLYP/SV(P),TZVP) and normalized UV-Vis (red, top) and CD (red, bottom) spectra of Ag(I)–(R3)-1(P). The blue sticks are computed electronic transitions that were subjected to Gaussian broadening (0.3 eV) and wavelength shift (−0.85 eV) to match the experimental spectra (black lines). (B) Computed electron density difference plots contributing to bands II–IV;38 the contour values are ±0.001 au. | |
Conclusions
This study describes our efforts focused on understanding the control of the chirality transfer in Ag(I)-folded molecular baskets. The results of both experimental and computational investigations are consistent, suggesting a structural interdependence within chiral baskets of type Ag(I)–(R3/S3)-1; a stereogenic alkyl center (with R or S configuration) at the “hinge” position can direct the twisting of pyridines at the rim to adopt either a clockwise P or counter-clockwise M orientation. A control of dynamic stereoisomerism in basket-like systems is important, and the results of our study will be useful for examining the relationship between chiral recognition and reactivity in gated and confined environments.
Acknowledgements
This work was financially supported with funds obtained from the National Science Foundation under CHE-1012146. Generous computational resources were provided by the Ohio Supercomputer Center. We thank Professor Jon R. Parquette of the Ohio State University for useful suggestions.
Notes and references
- J. M. Chance, J. H. Geiger and K. Mislow, J. Am. Chem. Soc., 1989, 111, 2326–2327 CrossRef CAS.
- R. Glaser, J. F. Blount and K. Mislow, J. Am. Chem. Soc., 1980, 102, 2777–2786 CrossRef CAS.
- H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175–182 CrossRef CAS.
- K. Mislow, D. Gust, P. Finocchiaro and R. J. Boettcher, Top. Curr. Chem., 1974, 47, 1–28 CAS.
- S. E. Gibson and M. P. Castaldi, Chem. Commun., 2006, 3045–3062 RSC.
- M. C. Keyes and W. B. Tolman, Adv. Catal. Processes, 1997, 2, 189–219 Search PubMed.
- P. Axe, S. D. Bull, M. G. Davidson, C. J. Gilfillan, M. D. Jones, D. E. J. E. Robinson, L. E. Turner and W. L. Mitchell, Org. Lett., 2007, 9, 223–226 CrossRef.
- M. Alajarin, C. Lopez-Leonardo, A. Vidal, J. Berna and J. W. Steed, Angew. Chem., Int. Ed., 2002, 41, 1205–1208 CrossRef CAS.
- J. W. Canary, C. S. Allen, J. M. Castagnetto and Y. Wang, J. Am. Chem. Soc., 1995, 117, 8484–8485 CrossRef CAS.
- Y. Yao, C. J. A. Daley, R. McDonald and S. H. Bergens, Organometallics, 1997, 16, 1890–1896 CrossRef CAS.
- G. Haberhauer, Angew. Chem., Int. Ed., 2008, 47, 3635–3638 CrossRef CAS.
- C. Moberg, Angew. Chem., Int. Ed., 2006, 45, 4721–4723 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- M. Von Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746–1757 CrossRef.
- R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 5119–5121 RSC.
- C. Moberg, Angew. Chem., Int. Ed., 1998, 37, 248–268 CrossRef CAS.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- J. W. Canary, S. Mortezaei and J. Liang, Coord. Chem. Rev., 2010, 254, 2249–2266 CrossRef CAS.
- S. Zahn and J. W. Canary, Science, 2000, 288, 1404–1406 CrossRef CAS.
- P. Axe, S. D. Bull, M. G. Davidson, M. D. Jones, D. E. J. E. Robinson, W. L. Mitchell and J. E. Warren, Dalton Trans., 2009, 10169–10171 RSC.
- B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS.
- M. Mikulas and K. Ruck-Braun, Org. Synth. Highlights IV, 2000, 187–193 Search PubMed.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280–2283 CrossRef CAS.
- L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef.
- J.-U. Rohde, M. R. Bukowski and L. Que, Curr. Opin. Chem. Biol., 2003, 7, 674–682 CrossRef CAS.
- A. I. Abouelatta, J. A. Sonk, M. M. Hammoud, D. M. Zurcher, J. J. McKamie, H. B. Schlegel and J. J. Kodanko, Inorg. Chem. (Washington, DC, U. S.), 2010, 49, 5202–5211 Search PubMed.
- Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC.
- D. J. Oh and K. H. Ahn, Org. Lett., 2008, 10, 3539–3542 CrossRef CAS.
- S. D. Bull, S. G. Davies, D. J. Fox, A. C. Garner and T. G. R. Sellers, Pure Appl. Chem., 1998, 70, 1501–1506 CrossRef CAS.
- G. Chelucci, S. Baldino, S. Chessa, G. A. Pinna and F. Soccolini, Tetrahedron: Asymmetry, 2006, 17, 3163–3169 CrossRef CAS.
- M. Gardlik, Z. Yan, S. Xia, S. Rieth, J. Gallucci, C. M. Hadad and J. D. Badjic, Tetrahedron, 2009, 65, 7213–7219 CrossRef CAS.
- S. Rieth, B.-Y. Wang, X. Bao and J. D. Badjic, Org. Lett., 2009, 11, 2495–2498 CrossRef CAS.
- S. Rieth, Z. Yan, S. Xia, M. Gardlik, A. Chow, G. Fraenkel, C. M. Hadad and J. D. Badjic, J. Org. Chem., 2008, 73, 5100–5109 CrossRef CAS.
- Z. Yan, S. Xia, M. Gardlik, W. Seo, V. Maslak, J. Gallucci, C. M. Hadad and J. D. Badjic, Org. Lett., 2007, 9, 2301–2304 CrossRef CAS.
- S. Rieth, X. Bao, B.-Y. Wang, C. M. Hadad and J. D. Badjic, J. Am. Chem. Soc., 2010, 132, 773–776 CrossRef CAS.
- S. Rieth, K. Hermann, B.-Y. Wang and J. D. Badjic, Chem. Soc. Rev., 2011 Search PubMed , in press.
- See Supplementary Information†.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 730–748 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- F. Kazmierczak, K. Gawronska, U. Rychlewska and J. Gawronski, Tetrahedron: Asymmetry, 1994, 5, 527–530 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- K. B. Wiberg, Acc. Chem. Res., 1999, 32, 922–929 CrossRef CAS.
- R. W. Hoffmann, Chem. Rev. (Washington, DC, U. S.), 1989, 89, 1841–1860 CrossRef CAS.
- J. Gawronski, F. Kazmierczak, K. Gawronska, U. Rychlewska, B. Norden and A. Holmen, J. Am. Chem. Soc., 1998, 120, 12083–12091 CrossRef CAS.
- R. B. Dyer, R. A. Palmer, R. G. Ghirardelli, J. S. Bradshaw and B. A. Jones, J. Am. Chem. Soc., 1987, 109, 4780–4786 CrossRef CAS.
- M. Claps, N. Parrinello, C. Saa, J. A. Varela, S. Caccamese and C. Rosini, Tetrahedron: Asymmetry, 2006, 17, 1387–1393 CrossRef CAS.
- S. Gladiali, G. Gottarelli, B. Samori and P. Palmieri, J. Chem. Soc., Perkin Trans. 2, 1980, 598–602 RSC.
- G. Gottarelli and B. Samori, Tetrahedron Lett., 1970, 2055–2058 CrossRef CAS.
- S. Grimme, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 361, 321–328 CrossRef CAS.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- C. S. Wilcox, Front. Supramol. Org. Chem. Photochem., 1991, 123–143 Search PubMed.
- C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524–2539 CrossRef CAS.
- Y.-G. Wang, J. Phys. Chem. A, 2009, 113, 10873–10879 CrossRef CAS.
-
K. Nakanishi, N. Berova, R. W. Woody and Editors, Circular Dichroism: Principles and Applications, 1994 Search PubMed.
-
D. A. Lightner and J. E. Gurst, Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy, 2000 Search PubMed.
- P. Skowronek, A. Katrusiak and J. Gawronski, Tetrahedron, 2002, 58, 10463–10468 CrossRef CAS.
- J. Gawronski and P. Skowronek, Curr. Org. Chem., 2004, 8, 65–82 CrossRef CAS.
- P. Mukhopadhyay, P. Wipf and D. N. Beratan, Acc. Chem. Res., 2009, 42, 809–819 CrossRef.
- P. Mukhopadhyay, G. Zuber, P. Wipf and D. N. Beratan, Angew. Chem., Int. Ed., 2007, 46, 6450–6452 CrossRef CAS.
- J. Neugebauer, Angew. Chem., Int. Ed., 2007, 46, 7738–7740 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, M. Levy, G. S. Painter and S. Wei, Phys. Rev. B, 1988, 37, 838–843 CrossRef CAS.
- J. W. Lockman, N. M. Paul and J. R. Parquette, Prog. Polym. Sci., 2005, 30, 423–452 CrossRef CAS.
- H. Shao and J. R. Parquette, Mol. Recognit. Polym., 2008, 259–306 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: additional experimental/computational results and spectra for new compounds. See DOI: 10.1039/c0sc00592d |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.