Unique reactivity of Grignard reagents towards cyclopropenyl-carboxylates—highly selective carbon–carbon bond cleavage†
Yu
Liu
a and
Shengming
Ma
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Process, Department of Chemistry, East China Normal University, 3663 North Zhongshan Lu, Shanghai, 200062, P. R. China
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Lu, Shanghai, 200032, P. R. China. E-mail: masm@sioc.ac.cn; Tel: +86 21 6260 9305
First published on 26th January 2011
Abstract
An unexpected highly selective reaction of Grignard reagents is described: Instead of generating the normal tertiary alcohols from the reaction with the ester functionality, its treatment with doubly activated cyclopropenyl-carboxylates leads to the deprotonation and highly regio- and stereoselective cleavage of the less substituted carbon–carbon single bond of the carbocycles with the Grignard reagent attacking the less substituted sp2carbon atom. Subsequent reaction with different electrophiles affords multi-substituted stereodefined 2-(1-alkenyl)malonate-type derivatives as single stereoisomers, which are otherwise very difficult to make.
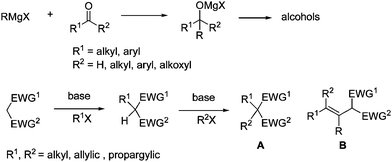
In organic chemistry, among many important transformations two reactions have dramatically changed the development of organic chemistry: one is the reaction of Grignard reagents with aldehydes/ketones and carboxylates yielding alcohols; the second one is the use of malonate-type of doubly activated methylene compounds connecting two fragments for the efficient construction of simple or complicated molecules for different purposes (Scheme 1). Thus, once there is an ester group in the molecule the use of Grignard reagents should be avoided unless this is the targeted functionality for reaction. In the second reaction, the synthesis of 2-(1-alkenyl)malonate-type active methine B is still challenging owing to the lower reactivity of 1-alkenyl halides towards malonate anions and the possible migration of the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond into conjugated compounds.1
C bond into conjugated compounds.1
 | ||
| Scheme 1 Two reactions that have changed the development of organic synthesis | ||
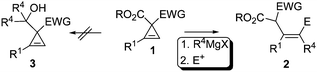
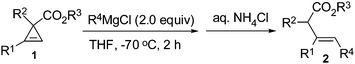
On the other hand, cyclopropene derivatives2 are readily available2c and highly strained,3 which make them easy to undergo the reactions with the carbon–carbon double bond4,5 or the selective cleavage of the two carbon–carbon single bonds.6,7 Thus, we envisioned that the reaction of such cyclopropenes with carbonucleophiles would provide a new type of stereodefined 2-(1-alkenyl)malonate derivative B (Scheme 1). However, there has been no success in this laboratory by trying with a series of different carbon nucleophilies such as mono- or two-electron-withdrawing-group-stabilized carbon anions. The well-known reactivity of Grignard reagents towards the ester group noted in Scheme 1 had deterred our study on the reaction of cyclopropenyl carboxylates with Grignard reagents.8 It is exciting to observe that when we finally tried this reaction, the results are surprising: no reaction occurred towards the ester functionality and the ring was opened. Here we wish to present the observed interesting deprotonation and highly regio- and stereoselective ring opening reaction of doubly activated cyclopropenes 1 with Grignard reagents leading to the formation of stereodefined multi-substituted olefins 2 with the R4group from the Grignard reagents attacking the less-substituted C-2 position of the cyclopropenes (Scheme 2).
 | ||
| Scheme 2 The reactions of cyclopropenes with Grignard reagents | ||
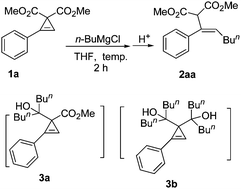
We firstly treated dimethyl 2-phenylcycloprop-2-ene-1,1-dicarboxylate 1a with n-butyl magnesium chloride. To our interest, instead of producing the alcohol 3a and the diols 3b, the Grignard reagent attacked the less-substituted sp2carbon atom of the cyclopropene ring, which was followed by protonolysis to give tri-substituted olefin 2aa as a single Z-isomer in a moderate yield (Table 1, entry 1). Higher loading of the Grignard reagents gave a better yield (Table 1, entries 1 to 4); conducting the reaction at a higher temperature (Table 1, entries 5 to 8) or in diethyl ether (Table 1, entry 9) didn't yield better results. Finally, we chose 2.0 equiv. of the Grignard reagent and THF as the solvent at −70 °C as the standard conditions.
Further investigation showed that this reaction enjoys a wide scope with the carboxylate group untouched (Table 2). Both 1°- and 2°-alkyl (entries 1, 3–6, 8, 14, 15, and 17) and phenyl (entries 2, 7, 9–13, and 16) Grignard reagents are applicable. Cyclopropenes with different R1 substituents such as aryl, Bn, or alkyl all yielded the products smoothly; again the acetoxy group remains intact: the reaction occurred exclusively on the cyclopropene ring (entry 14). Four equivalents of Grignard reagents were required for the reaction at −30 °C to ensure a higher yield when one of the activating groups is SO2Ph (entries 6, 7, and 14). The reaction of unsubstituted dimethyl cycloprop-2-ene-1,1-dicarboxylate 1j also proceeded smoothly with alkyl (entries 15 and 17) or phenyl (entry 16) Grignard reagents in excellent stereoselectivity to afford the E-isomers as single products although the yields are relatively lower.
| Entry | 1 | R4 | Isolated yield (%) | ||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| a 34% of 1a was recovered. b 4.0 equiv. of Grignard reagent were added. c The reaction was conducted at −30 °C. d 3.0 equiv. of Grignard reagent were added. | |||||
| 1 | Ph | CO2Me | Me (1a) | n-Bu | 66 (2aa) |
| 2 | Ph | CO2Me | Me (1a) | Ph | 77 (2ab) |
| 3 | Ph | CO2Me | Me (1a) | Et | 64 (2ac) |
| 4 | Ph | CO2Me | Me (1a) | i-Pr | 61 (2ad) |
| 5a | Ph | CO2Me | Me (1a) | Cy | 51 (2ae) |
| 6b,c | Ph | SO2Ph | Et (1b) | n-Bu | 54 (2ba) |
| 7b,c | Ph | SO2Ph | Et (1b) | Ph | 50 (2bb) |
| 8 | p-ClC6H4 | CO2Me | Me (1c) | n-Bu | 62 (2ca) |
| 9 | p-MeC6H4 | CO2Me | Me (1d) | Ph | 82 (2db) |
| 10d | p-O2NC6H4 | CO2Me | Me (1e) | Ph | 78 (2eb) |
| 11 | Bn | CO2Me | Me (1f) | Ph | 73 (2fb) |
| 12 | n-Bu | CO2Me | Me (1g) | Ph | 76 (2gb) |
| 13 | n-C8H17 | CO2Me | Me (1h) | Ph | 81 (2hb) |
| 14b | -(CH2)2OAc | SO2Ph | Et (1i) | n-Bu | 46 (2ia) |
| 15 | H | CO2Me | Me (1j) | n-Bu | 55 (2ja) |
| 16 | H | CO2Me | Me (1j) | Ph | 51 (2jb) |
| 17 | H | CO2Me | Me (1j) | Et | 47 (2jc) |
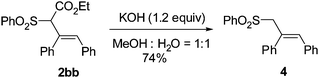
In order to further confirm the stereoselectivity, the Z-product 2bb was treated with KOH to yield the decarboxylation Z-product 4 in 74% yield (eqn (1)). The X-ray diffraction study of this compound further confirmed the Z-configuration of the CC bond in 2 (Fig. 1).9
 | (1) |
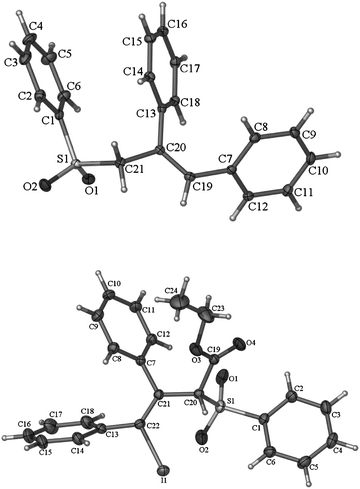 | ||
| Fig. 1 The X-ray diffraction of 4 (up) and 5c (down). | ||
More excitingly, it is a surprise for us to note that the treatment of the in situ formed intermediate, generated from the reaction of 1b and PhMgCl under this optimized conditions, with AcOD afforded di-deuterated product 2bb-d. The deuterations at both positions are ≥92% as judged by the 1H NMR analysis of the crude product. However, the deuteration at the sp3carbon atom decreased to 71% after chromatographic purification on silica gel, which was caused by the smooth exchange of this acidic D atom and the H atom from the environment. This result proved the formation of unexpected C1-type intermediate indicating that two groups may be introduced to the non-substituted sp2carbon atom in cyclopropenes 2 (Scheme 3).
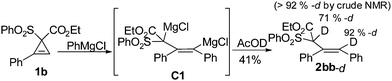 | ||
| Scheme 3 The reaction of cyclopropene 1b with PhMgCl quenched with DOAc | ||
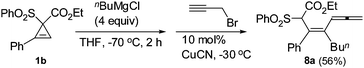
To further demonstrate the potential of this unique transformation, the in situ generated C1-type intermediates from 1a or 1b were quenched with different electrophiles (Table 3): the reaction with I2 gave alkenyl iodides as single stereoisomers 5a–c (entries 1, 4 and 7), which are complementary to the corresponding E-isomers formed by the reaction with inorganic iodides with the regioselectivity nicely addressed (Scheme 2).7,10 The structure of the product 5c was unambiguously established by X-ray diffraction study (Fig. 1).11 The intermediates may also be trapped by a range of allyl halides (entries 2, 5, 8, 9 and 11), methyl iodide (entries 3, 6 and 10), and propargyl bromide(eqn (2), yielding allene 8a)12 with CuCN as the catalyst.
 | (2) |
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 mol% of CuCN was added as the catalyst. bIn some cases products 5–7 were contaminated with corresponding protonolysis products. 10 mol% of CuCN was added as the catalyst. bIn some cases products 5–7 were contaminated with corresponding protonolysis products. |
|---|

|
Based on the above study we propose the following mechanism (Scheme 4): The Grignard reagent firstly reacts with cyclopropene to deprotonate the olefinic proton forming cyclopropenyl magnesium D.13 Interestingly, it is observed that the intermediate D did not undergo the isomerization forming propargylic anion as noted by Fox et al.5c probably due to the unique property of the magnesium reagent and the presence of the excess Grignard reagent leading to the observed ring-opening reaction. The bidentate coordination of the ester groups with the magnesium7 may also contribute to the stabilization of the intermediate, and further promoted the subsequent highly regioselective attack of the Grignard reagent at the negatively charged sp2carbon atom in D forming dimagnesium intermediate C. This intermediate gives the product after quenching with different electrophiles. Through this reaction, the cyclopropene derivatives 1 may be considered as the E-type synthon potentially useful for the efficient construction of stereodefined highly functionalized olefins 2 with high regio- and stereoselectivity.
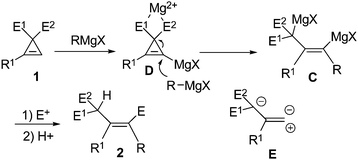 | ||
| Scheme 4 Plausible mechanism. | ||
To further develop the potentials of this reaction, the diene 6a has been treated with NaH to introduce allyl, propargyl, and methyl groups by reacting with different organic halides. Thus, the corresponding triene 9c could be transformed into cycloheptadiene 10 in 82% yield via a RCM reaction (Scheme 5).
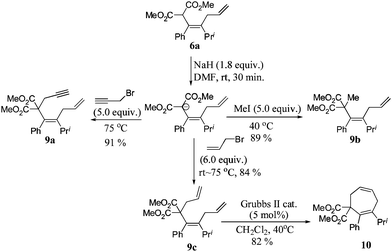 | ||
| Scheme 5 Further application of the product. | ||
In conclusion, we observed the unique reactivity of Grignard reagents towards the cyclopropene dicarboxylates: no reaction was observed to the ester group. The observed novel deprotonation and highly regio- and stereoselective carbon–carbon bond cleavage reaction of cyclopropenes with Grignard reagents leads to stereodefined multi-substituted olefins with high loading of functionality, which are otherwise difficult to make. The reaction provides a wide range of synthetically attractive C-type dimagnesium intermediates from the readily available cyclopropenes, Grignard reagents, and electrophiles. Further studies on this reaction are being pursued in our laboratory.
Financial support from the State Basic Research & Development Program of China (NO. 2009CB825300) and National Natural Science Foundation of China (20732005) is greatly appreciated. We thank Mr. W. Yuan in this group for reproducing the results presented in entries 5, 9 and 16 in Table 1.
Notes and references
- For preparation of β,γ-unsaturated esters from phenylacetaldehyde, see: (a) T. R. Hoye and W. S. Richardson, J. Org. Chem., 1989, 54, 688 CrossRef CAS. For copper-catalyzed coupling of malonate esters with vinyl bromides, see: (b) L. Pei and W. Qian, Synlett, 2006, 1719 CAS.
- For some recent reviews on cyclopropenes, see: (a) M. S. Baird, Chem. Rev., 2003, 103, 1271 CrossRef CAS; (b) J. M. Fox and N. Yan, Curr. Org. Chem., 2005, 9, 719–732 CrossRef CAS; (c) M. Rubin and V. Gevorgyan, Synthesis, 2006, 8, 1221; (d) M. Rubin and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS; (e) I. Marek, S. Simman and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364–7376 CrossRef CAS.
- (a) R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2006, 128, 4598 CrossRef CAS; (b) A. Fattahi, R. E. McCarthy, M. R. Ahmad and S. R. Kass, J. Am. Chem. Soc., 2003, 125, 11746 CrossRef CAS.
- For selected most recent references on the reactions occurred to the CC bonds, see:
(a) W. M. Sherill and M. Rubin, J. Am. Chem. Soc., 2008, 130, 13804 CrossRef;
(b) B. K. Alnasleh, W. M. Sherrill and M. Rubin, Org. Lett., 2008, 10, 3231 CrossRef CAS;
(c) J. Yin and J. D. Chisholm, Chem. Commun., 2006, 632 RSC;
(d) E. A. F. Fordyce, T. Luebbers and H. W. Lam, Org. Lett., 2008, 10, 3993 CrossRef CAS.
- For transition metal-catalyzed functionalization of cyclopropenes, see: (a) S. Unteidt and A. de Meijere, Chem. Ber., 1994, 127, 1511 CAS; (b) W. Xu and Q.-Y. Chen, J. Org. Chem., 2002, 67, 9421 CrossRef CAS; (c) L. Liao, N. Yan and J. M. Fox, Org. Lett., 2004, 6, 4937 CrossRef CAS; (d) S. Chuprakov, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 3714 CrossRef CAS; (e) S. Chuprakov, D. A. Malyshev, A. Trofimov and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 14868 CrossRef CAS; (f) E. A. F. Fordyce, Y. Wang, T. Luebbers and H. W. Lam, Chem. Commun., 2008, 1124 RSC.
- For selected reports on the ring-opening reactions of cyclopropenes see: (a) P. Binger and H. Schäfer, Tetrahedron Lett., 1975, 4673 CrossRef CAS; (b) H. G. Richey Jr., B. Kubala and M. A. Smith, Tetrahedron Lett., 1981, 22, 3471 CrossRef; (c) I. Nakamura, G. B. Bajracharya and Y. Yamamoto, J. Org. Chem., 2003, 68, 2297 CrossRef CAS; (d) M. A. Smith and H. G. Richey Jr., Organometallics, 2007, 26, 609 CrossRef CAS; (e) Y. Wang, E. A. F. Fordyce, F. Y. Chen and H. W. Lam, Angew. Chem., Int. Ed., 2008, 47, 7350 CrossRef.
- (a) S. Ma, J. Zhang, Y. Cai and L. Lu, J. Am. Chem. Soc., 2003, 125, 13954 CrossRef CAS; (b) S. Ma, J. Zhang, L. Lu, X. Jin, Y. Cai and H. Hou, Chem. Commun., 2005, 909 RSC; (c) Y. Wang and H. Lam, J. Org. Chem., 2009, 74, 1353 CrossRef CAS.
- For some recent reports on reactions of cyclopropenylmethanols with organomagnesiums, see: (a) L. Liao and J. M. Fox, J. Am. Chem. Soc., 2002, 124, 14322 CrossRef CAS; (b) X. Liu and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 5600 CrossRef CAS; (c) Z. Yang, X. Xie and J. M. Fox, Angew. Chem., Int. Ed., 2006, 45, 3960 CrossRef CAS; (d) S. Simaan, A. Marasawa, P. Bertus and I. Marek, Angew. Chem., Int. Ed., 2006, 45, 3963 CrossRef CAS; (e) N. Yan, X. Liu and J. M. Fox, J. Org. Chem., 2008, 73, 563 CrossRef CAS; (f) X. Xie, Z. Yang and J. M. Fox, J. Org. Chem., 2010, 75, 3847 CrossRef CAS ; For a report on the similar reaction of cyclopropenone acetals, see: M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 2000, 122, 978 Search PubMed ; For reviews, see: 2c and 2e.
- Crystal data for 4: C21H18O2S, Mr = 334.41, monoclinic, space groupP21, final R indices [I > 2σ(I)], R1 = 0.0316, wR2 = 0.0831, R indices (all 13.3325(6) Å, α = 90°, β = 102.4790(10)°, γ = 90°, V = 857.51(7) Å3, T = 173(2) K, Z = 2, reflections collected/unique: 9963/3027 (Rint = 0.0247), number of observations [>2σ(I)] 2977, parameters: 218. CCDC 771437.
- The following reaction would provide a regioisomeric mixture:
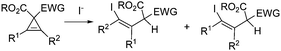 .
. -
Crystal data for 5c: C48H41I2O8S2, Mr = 1063.73, triclinic, space groupP
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , final R indices [I > 2σ(I)], R1 = 0.0262, wR2 = 0.0667, R indices (all data) R1 = 0.0322, wR2 = 0.0835, a = 11.6322(6) Å, b = 5.6630(3) Å, c = data) R1 = 0.0298, wR2 = 0.0703, a = 10.4567(3) Å, b = 14.5947(4) Å, c = 16.0710(4) Å, α = 82.1840(10)°, β = 80.0170(10)°, γ = 70.3230(10)°, V = 2266.47(11) Å3, T = 296(2) K, Z = 2, reflections collected/unique: 26417/7945 (Rint = 0.0164), number of observations [>2σ(I)] 7211, parameters: 554. CCDC 771438.
, final R indices [I > 2σ(I)], R1 = 0.0262, wR2 = 0.0667, R indices (all data) R1 = 0.0322, wR2 = 0.0835, a = 11.6322(6) Å, b = 5.6630(3) Å, c = data) R1 = 0.0298, wR2 = 0.0703, a = 10.4567(3) Å, b = 14.5947(4) Å, c = 16.0710(4) Å, α = 82.1840(10)°, β = 80.0170(10)°, γ = 70.3230(10)°, V = 2266.47(11) Å3, T = 296(2) K, Z = 2, reflections collected/unique: 26417/7945 (Rint = 0.0164), number of observations [>2σ(I)] 7211, parameters: 554. CCDC 771438. - For monographs and books on allenes: (a) S. R. Landor, The Chemistry of Allenes, Academic, London, 1982 Search PubMed; (b) N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) N. Krause, Cumulenes and Allenes in Science of Synthesis, Vol. 44, Thieme, Stuttgart, 2008 Search PubMed. For some recent reviews: (d) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef; (e) B. Alcaide and P. Almendros, Eur. J. Org. Chem., 2004, 3377 CrossRef CAS; (f) S. Ma, Aldrichimica Acta, 2007, 40, 91 CAS; (g) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef; (h) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS.
- For selected examples on deprotonation of cyclopropenes, see: (a) A. M. Moiseenkov, B. A. Czeskis, T. Y. Rudashevskaya, O. A. Nesmeyanova and A. V. Semenovskyf, Tetrahedron Lett., 1981, 22, 151 CrossRef CAS; (b) K. Sorger, P. von Rague Schleyer and D. Stalke, J. Chem. Soc., Chem. Commun., 1995, 2279 RSC; (c) M. Nakamura, H. Isobe and E. Nakamura, Chem. Rev., 2003, 103, 1295 CrossRef CAS and references therein; (d) I. Zrinski, G. Gadanji and M. Eckert-Maksic, New J. Chem., 2003, 27, 1270 RSC; (e) I. Zrinski and M. Eckert-Maksic, Synth. Commun., 2003, 33, 4071 CrossRef CAS; (f) I. Zrinski, N. Novak-Coumbassa and M. Eckert-Maksic, Organometallics, 2004, 23, 2806 CrossRef; (g) L. A. Fisher and J. M. Fox, J. Org. Chem., 2008, 73, 8474 CrossRef CAS ; see also refs. 5a and 5c.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures, analytical data, and 1H/13C NMR spectra. CCDC reference numbers 771437–771438. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00584c |
| This journal is © The Royal Society of Chemistry 2011 |