Constitutionally selective amplification of multicomponent 84-membered macrocyclic hosts for (−)-cytidine•H+†
Mee-Kyung
Chung
a,
Kay
Severin
b,
Stephen J.
Lee
c,
Marcey L.
Waters
a and
Michel R.
Gagné
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: mgagne@unc.edu; Tel: (+1)919 962 6341
bInstitut des Sciences et Ingénieries Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland
cUS Army Research Office, P.O. Box 12211, Research Triangle Park, NC 27709, USA
First published on 24th January 2011
Abstract
Mixtures of dipeptide monomers create stereochemically and constitutionally complex dynamic libraries of potential receptors. When (−)-cytidine was utilized as guest an 84-membered cyclic host was amplified (70–175 fold) from a nearly undetectable initial concentration. Only the specified diastereomeric combination of the two chiral building blocks yielded a dynamic library from which the macrocyclic receptor could be amplified.
The templating of dynamic constitutional libraries has developed into a powerful tool for discovering synthetic receptors.1 Stereochemically and structurally diverse libraries have led to both enantio- and diastereoselective receptors for a broad array of important analytes.1,2 The minimization of entropic costs provides a speciation force for single component (homo) libraries that usually favors relatively small, conformationally constrained, macrocyclic hosts. In the case of hetero (multicomponent) libraries, the growth in the number of possible constitutional isomers can mitigate some of the entropic cost of larger structures. For example in a two component library, 13 unique non-catenated cyclic hexamers can be assembled. By contrast, a homo library can only assemble 1 macrocyclic hexamer.3
From the perspective of a dynamic library of molecular receptors, the free energy balance in macrocyclic host–guest complexes generally, but not exclusively, lies towards hosts with 4 or less units.1,2 This is especially true of homolibraries (see above argument). In as much as larger structures might also be floppier, the selection of such species in a competitive binding assay suggests that the macrocycles are either unusually preorganized2f or are able to efficiently collapse onto the guest (e.g. induced fit receptors).2g
We have previously demonstrated that templating of racemic dynamic libraries (DLs) with chiral guests can be used to rapidly identify enantioselective receptors.2d,4 Described herein are highly amplified hexameric macrocyclic receptors for (−)-cytidine•H+ that are composed of a constitutionally precise arrangement of four units of monomer 1 and two units of 2. In the absence of (−)-cytidine, the equilibrium amounts of these 84-membered cyclic hosts is nearly undetectable, demonstrating that strong host–guest interactions can overcome inherently low speciation tendencies.
When pairs of D- and L-proline-based dipeptide monomers (D-1 and L,L-2a,b) were treated with trifluoroacetic acid (TFA), a complex mixture of cyclic oligomers was obtained through hydrazone exchange (Scheme 1). This library included cyclic dimers, trimers, tetramers, along with traces of pentamers and hexamers, as well as [2]-catenated octamers in the case of 2b;5 multiple constitutional isomers of each n-mer were observed.6
 | ||
| Scheme 1 DLs from D-1 and L,L-2a or b. Note that the monomers differ from the oligomer repeat unit by two CH3OH units. | ||
When the DL formed from D-1 and L,L-2a was templated with (−)-cytidine (3 eq. with respect to total monomers), a nearly undetectable cyclic hexamer (3a) was amplified ∼70 fold.7 At equilibrium, this species accounted for 14% of the total monomers as determined by HPLC-UV. For the combination D-1 and L,L-2b, the corresponding cyclic hexamer 3b was even more highly amplified (46% of the total monomers, AF = 175 ± 25) (Fig. 1).7,8
 | ||
Fig. 1 HPLC-UV trace (289 nm) at equilibrium (day 30) of untemplated (blue) and (−)-cytidine templated (red) DLs from D-1 and L,L-2b (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5.0 mM in MeCN–CHCl3 = 1:3). The y-axis is normalized. :1, 5.0 mM in MeCN–CHCl3 = 1:3). The y-axis is normalized. | ||
Although not fully investigated, exploratory (−)-cytidine templating experiments using 2 monomers with R = iBu, p-CF3Ph, 1-naphthyl or 2-naphthyl, similarly led to the amplification of a single hexamer (ESI Fig. 2†), whereas DLs with R = Bn (Phe), CH2CH2SMe (Met), CH2-indole (Trp) or CHOHMe (Thr) did not.
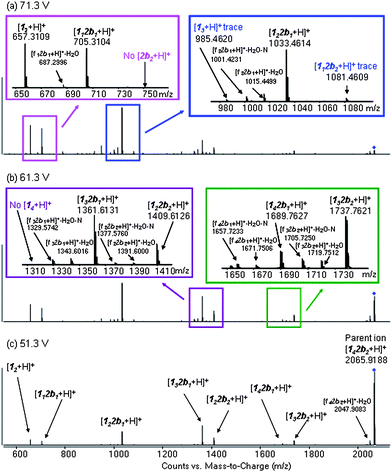 | ||
| Fig. 2 LC-QTOF (CID MS/MS) spectra of the isolated 3b at a collision energy of (a) 71.3 V, (b) 61.3 V and (c) 51.3 V. The y-axis is normalized. | ||
LC-MS analysis revealed that 3a and 3b were assembled from four units of 1 and two of 2a or 2b (ESI Fig. 5†). Fragmentation into pentamers through dimers in the MS/MS of isolated 3b (Fig. 2) excluded a [2]-catenane type structure (i in Scheme 2).2f,5 The lack of [2b2b2 + H]+ dimeric and [114 + H]+ tetrameric daughter ions also excluded the “ortho” isomer (ii in Scheme 2).
 | ||
| Scheme 2 Possible daughter ions for the isomers of 3b. | ||
The observation of [1122b2b1 + H]+ as the dominant trimeric daughter ion and the absence of [2b2b2 + H]+ suggested that 3b was the symmetric constitutional isomer with alternating 12 and 2b units, i.e., cyclo [1•1•2b•1•1•2b] rather than the “meta” isomer. As shown in Fig. 2a, traces of [113 + H]+ and [1112b2b2 + H]+ could be detected. Although it is difficult to eliminate the possibility that traces of a second isomer is present in the sample, we have previously noted that hydrazones are sensitive to ion-ion crossover in the mass spectrometer.9
A second 142b2 isomer of 3b was observed in an LC-QTOF (MS/MS) analysis of the templated library. The presence of three trimeric daughter ions ([113 + H]+, [1122b2b1 + H]+ and [1112b2b2 + H]+), two dimeric ions, two tetrameric ions, and two pentameric daughter ions (Fig. 3) combined with the lack of [2b2b2 + H]+ and [114 + H]+ daughter ions, implied that this minor isomer of 3b was the “meta” isomer (iii in Scheme 2). The unique MS signatures of this minor isomer suggests that ion fragmentation analysis can be used to assign constitutional isomer of pure samples.9
 | ||
| Fig. 3 A portion of LC-QTOF UV trace (289 nm) of 3b and its isomer (blue circle) in (−)-cytidine templated DLs from D-1 and L,L-2b and LC-QTOF (CID MS/MS) spectra of the isomer of 3b at a collision energy of (a) 71.3 V, (b) 61.3 V and (c) 51.3 V. The y-axis is normalized. | ||
1H NMR analysis of 3b isolated by semi-preparative HPLC was informative and confirmed the “para” assignment suggested by MS/MS. Despite the large size of the macrocycle, the 1H NMR spectra was surprisingly sharp suggesting that 3b existed in one predominant conformation or that conformational exchange was rapid on the NMR time scale. Taking into account the directionality of each unit (represented by an arrow), symmetry arguments suggested that the “para” isomer could be distinguished from the “ortho” and “meta” by determining the number of AIB methyl groups.

Both the ortho and meta isomers have eight inequivalent CH3 groups, while the C2 symmetric para isomer should only lead to four. As shown in Fig. 4, the 1H NMR spectrum in pyridine-d5 was sharp and four unique CH3 groups were observed. Additionally supportive was the observation of a single phenylglycine CHαgroup and its adjacent (coupled; COSY, ESI Fig. 8†) amide NH group.
 | ||
| Fig. 4 599.8 MHz 1H NMR spectra of 3b at 20 °C; (a) in pyridine-d5, (b) a portion (δ = 1.3 to 2.7) of the 1H NMR spectra showing the 4 AIB methyl groups that are only consistent with the “para” isomer. | ||
The proline portion of the TOCSY spectrum showed 3b to contain three (not six) inequivalent proline units, additionally consistent with a C2-symmetric structure (Fig. 5). MS and NMR data collectively thus show that (−)-cytidine•H+ selects the C2-“para” constitutional isomer.
 | ||
| Fig. 5 A portion (δ = 1.5 to 4.9) of the 599.8 MHz TOCSY spectrum of 3b at 20 °C in pyridine-d5. Three unique proline residues are observed. | ||
Although it would be preferable to directly measure the binding constant, (−)-cytidine is protonated under the DL conditions and we have been unable to discover conditions wherein (−)-cytidine•H+ is soluble and the library does not begin to reequilibrate under the acidic condition.10 To overcome this limitation we have resorted to numerical simulation11 methods to estimate host–guest binding. Equilibrium speciations for a DL of D-1 + L,L-2a (1:1, 5.0 mM total) were collected as a function of (−)-cytidine concentration (1.25–15 mM; see ESI†). This data was fit using the program DCLFit11d to a model that assumes each of the 25 macrocycles12 present in solution can form a 1:1 adduct with the template (larger oligomers were ignored for the calculations).13 Good agreement between the data and the model was obtained (ESI Fig. 9†). The calculations showed most species to be weak binders (−10 to −15 kJ mol−1), with 3a standing out (−21.0 kJ mol−1).14
Since 3a and 3b were composed of two chiral building blocks, the effect of monomer stereochemistry on the chiral recognition was tested. All pair-wise stereochemical combinations of 1, 2a and 2b were examined and only D-1/L,L-2a or D-1/L,L-2b led to hexamer amplification.15 In as much as amplification reflects a competitive binding affinity, the molecular recognition of (−)-cytidine•H+ by 3 is enantioselective, as the enantiomer of 3 is not amplified under the same conditions. While only a small subset of the possible diastereomers of 3 were accessible via the pairwise combination of monomers (total 8 combinations), 3 was found to be the only viable hexameric host, suggesting that subtle (or perhaps not) conformational changes are not tolerated in the ternary [3•(−)-cytidine•H+] complex.
In conclusion, we have identified complex, stereochemically and constitutionally precise macrocycles of unusually large size that bind (−)-cytidine•H+. Although precise binding constants were not obtainable, the magnitude of the amplification factors and the simulations pointed to high affinities for these 84-membered macrocycles, and reaffirm the notion that high entropic costs can be overcome in receptor assembly.
Acknowledgements
M.R.G. and M.L.W. thank the Defense Threat Reduction Agency (DTRA) for support (W911NF04D0004), S.J.L. thanks the Army Research Office, and K.S. thanks the Swiss National Science Foundation and EPFL for kind support.Notes and references
- For reviews, see:
(a)
J. J. Becker, M. R. Gagné, Chiral selection in DCC. In Dynamic Combinatorial Chemistry: In Drug Discovery, Bioorganic Chemistry and Material Science; Miller, B. C., ed.; Wiley: Hoboken, NJ, 2010; pp 155 Search PubMed
; (b) S. Ladame, Org. Biomol. Chem., 2008, 6, 219 RSC
-
(a) M. Hutin, C. J. Cramer, L. Gagliardi, A. R. M. Shahi, G. Bernardinelli, R. Cerny and J. R. Nitschke, J. Am. Chem. Soc., 2007, 129, 8774 CrossRef CAS
- K. Severin, Chem. Eur. J., 2004, 10, 2565 CrossRef CAS
- M.–K. Chung, C. R. Hebling, J. W. Jorgenson, K. Severin, S. J. Lee and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 11819 CrossRef CAS
- M.–K. Chung, P. S. White, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 8683 CrossRef CAS
- We estimate a total of 34 non-catenanted species between dimers and hexamers.
- AF = [3a]templated/[3a]untemplated; a precise value for AF is difficult to obtain due to the inaccuracy in determining [3a]untemplated (≤ 10 μM at equilibrium). See ESI Fig. 1†.
- For a similar consumption of catenane upon guest addition, see: K. R. West, R. F. Ludlow, P. T. Corbett, P. Besenius, F. M. Mansfeld, P. A. G. Cormack, D. C. Sherrington, J. M. Goodman, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2008, 130, 10834 Search PubMed
- Ion-ion reactions of hydrazone mixtures can lead to ions that are an artifact of crossover. LC-separation prior to analysis significantly attenuates these problems: H. Schiltz, M.–K. Chung, S. J. Lee and M. R. Gagné, Org. Biomol. Chem., 2008, 6, 3597 Search PubMed
- Isolated host was found to reequilibrate with even weaker acids (e.g.CH3COOH).
- For numerical simulations of DCLs see ref. 4 and:
(a) R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2010, 132, 5984 CrossRef CAS
- The simulation was simplified by combining the low binding constitutional isomers of meso-tetramer (122a2) and the hetero-pentamers (132a2, 122a2) and hexamers (122a4, 132a3).
- The size of this dataset precluded a modelling using the Gepasi method discussed in references 3, 4, and 11g.
- While there were two other receptors (15 and 152a1) with high binding constants (−22 kJ mol−1 and −18 kJ mol−1, respectively), these species were present in very low concentrations upon templating. Although the binding affinity of homo-pentamer (15) is similar to that of the hexamer, it is known that homo-aggregates are disadvantaged over hetero-aggregates due to mass balance considerations (competition for limited resources). For a detailed discussion, see: ref. 2h and 3.
- None of the combination of D-1/L,D-2x; D-1/D,L-2x; D-1/D,D-2x; L-1/L,D-2x; L-1/D,L-2x; L-1/D,D-2x; L-1/L,L-2x (x = a or b) led to the amplification of a cyclic hexamer. Not tested were DLs containing multiple combinations of monomer 2, which would access many additional diastereomers of the cyclic hexamer. A potential complication in the broader interpretation of these results was the observation of a library precipitate when L,D- or D,L- isomers of 2 were tested. In each case, however, significant concentration of library remained in the supernatant and its speciation (by LC-MS) was nearly identical to non-templated experiments (ESI Fig. 11 and 12†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the DCLs, LC-MS and CID-MS/MS analyses of templated DCLs, simulation details and speciation data. See DOI: 10.1039/c0sc00548g |
| This journal is © The Royal Society of Chemistry 2011 |