Mechanised materials
Megan M.
Boyle
a,
Ronald A.
Smaldone
a,
Adam C.
Whalley
a,
Michael W.
Ambrogio
a,
Youssry Y.
Botros
abc and
J. Fraser
Stoddart
*a
aDepartment of Chemistry and Department of Materials Science and Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA. E-mail: stoddart@northwestern.edu
bIntel Labs, Building RNB-6-61, 2200 Mission College Blvd, Santa Clara, CA 95054-1549, USA
cNational Center for Nano Technology Research, King Abdulaziz City for Science and Technology (KACST), P.O. Box 6086, Riyadh, 11442, Kingdom of Saudi Arabia
First published on 16th December 2010
Abstract
Throughout history, mankind has attempted to mimic the natural world—building wings to fly like the birds and fabricating systems to harness energy from the sun like our botanical brethren. However, on account of the enormous complexity that Nature has accrued over millions of years—more often than not—this imitation has resulted in failure. It was only once systems were developed that sought a looser connection to Nature, rather than attempting to reproduce it in a direct manner, that we were successful in accomplishing our goals. It is tempting to take these lessons we have learned from the macroscopic world and apply them to the nanoscale. Rather than attempting to replicate biological molecular machines capable of performing macroscopic motion, it is important to create a more robust world. In the past two decades, chemists have been synthesizing molecular switches and assembling molecular machines in order to study their properties and understand their function. Just as the artist takes to his or her canvas to create some masterpiece, chemists need to move to surfaces and interfaces in order to illicit function from these advanced integrated systems. In this mini-review we (1) outline several synthetic molecular machines that have been pinned down on surfaces to induce macroscale function and motion and (2) highlight some bioconjugated molecular devices which are capable of harnessing motion. Finally, we reflect upon the concept of appending molecular machinery to biological entities in order to express a range of properties.
Introduction
Nature is populated with a plethora of complex integrated systems which harness motion on the nanometre scale to elicit macroscale function through highly coordinated and organized conformational changes and supramolecular control. Some of Nature's most remarkable molecular machines rely on the performance of motor proteins like myosin,1kinesin,2 and the adenosine triphosphate (ATP)-powered F1-ATPase3 for their operation. In myosin, for example, chemical energy (ATP) is converted into mechanical energy when the motor protein walks along linear bundles of another protein, namely actin. The elongations and contractions that result from the molecular motion of myosin are amplified such that they translate into useful macroscale motion, allowing for the performance of essential biological functions, e.g., muscle contraction, cell division, transport of membrane vesicles, and cell locomotion.4 Bioconjugate chemistry utilises the movements that biological machines perform in order to develop hybrid systems, ranging from molecular motors that inhibit enzymatic activity,5 to vehicles capable of transporting unnatural cargos.6 A major challenge which still remains, however, is the development of wholly synthetic integrated systems capable of converting nanoscale movements into macroscopic motion. As chemical scientists begin to realise that there is more to chemistry than simply trying to mimic the machinery of biological systems—using unnatural products and protocols to yield imitations of sometimes questionable value and significance—we will start to focus our efforts on the design and production of advanced integrated systems capable of carrying out prescribed functions of a more and more sophisticated type.Chemists have come a long way in the past two decades towards developing a diverse range of artificial nanoscale machines capable of correlated motion.7 These molecular machines range in complexity from molecules which demonstrate unidirectional rotational motion8 to mechanically interlocked molecular switches9 and even more complex devices which rely upon switches assembled on surfaces10 and in tunnel junctions11 for their performance. Furthermore, biomolecules have been enhanced with artificial machines/switches which endow them with precisely controlled properties.5,6
The message that comes over so far from most of these successes recorded—upon taking operating molecular machinery out of solution,8 onto surfaces,10 and into interfaces11—is that robustness seems to be a requirement for progress to be made in the context of complex integrated systems which incorporate molecular machines and switches.12 The situation we face in the wake of the systems that have demonstrated some modicum of promise could be likened to our eternal drive as human beings to take to the air in a practical manner. Thus, today's passenger jet airliners are robust flying machines that have no more than a passing resemblance to the birds and the bees and the bats. Hence, it is not unlikely, at least in the present phase of development, that switchable mechanically interlocked molecules13 (MIMs) with machine-like characteristics will find their way into robust settings like metal–organic frameworks14 (MOFs) where they can eventually exhibit macroscale movement and function.15
Synthetic molecular machines
In order to achieve the goal of truly functional devices derived from artificial molecular machinery, our synthetic methodology must be married to robust, structured materials capable of providing order and direction. Molecular scale motion, in the form of translation or rotation, can certainly be generated. However, nanoscale or macroscale movement, resulting from molecular activity, has proven much more challenging to realise. In this section, we will review briefly some of the successes of introducing artificial molecular motors onto 2D surfaces before moving on to discuss more complex 3D architectures. We will focus on the mechanism of molecular switching and the functions that can be elicited.A wide range of molecular rotors, capable of converting light and chemical energy into motion have been synthesised8 and their properties in solution have been thoroughly explored.16 In order to harness the power of such systems, however, it is necessary to locate these machines at surfaces, such that the work can be converted into useful energy. In the literature, this conversion has been accomplished using a number of different systems.17 One such example utilises chiral helical alkenes, which were synthesised and attached to gold nanoparticles (Fig. 1a).18 The rotors were attached to the gold surface by two tethers to prevent undesired thermal rotation of the system. The required rotary motion can be achieved by irradiating the surface with UV light—thus taking advantage of two energetically uphill photo-isomerisations, followed by two irreversible helix inversions. This process drives one half of the rotor to rotate a full 360° with respect to the other half. Furthermore, as a result of the chirality of the molecule and the irreversibility of the helix inversion, the rotor rotates in one, and only one, direction. In another system,19 a light-driven molecular motor was doped into a cholesteric liquid crystalline film (Fig. 1b). As a result of the photoswitch, irradiation of the film causes a change in the helicity of the liquid crystalline phase. This rotation can be harnessed to rotate large objects placed on the surface, such as a microscopic glass rod.
 | ||
| Fig. 1 (a) An example of a molecular motor tethered to a gold surface.18 The rotor is connected to a stator by a carbon–carbon double bond—the axis of rotation. The stator is tethered to the surface by two legs in order to ensure rotation is not the product of uncontrolled motion. Irradiation with UV light causes photoisomerisation such that the methyl substituent assumes an energetically disfavoured pseudo-equatorial orientation (Step 1). Thermal energy can be used to cause an irreversible helix inversion (Step 2), resulting in a net rotation of 180°. In steps 3–4, the process is repeated to yield a net rotation of 360°. (b) A similar molecular rotor, which is attached to the stator by a carbon–carbon double bond, can be used as a dopant in a cholesteric liquid-crystal (LC) film.19 Irradiation with UV light causes rotation of the film such that removal of the light source elicits rotation in the opposite direction, causing a microscopic glass rod on the surface of the LC film to rotate. Scale bars = 50 μm. | ||
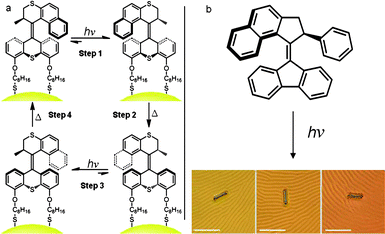
Deformation of a robust material was achieved by coating a surface with responsive molecules. Palindromic bistable [3]rotaxanes, applied to a thin gold surface, can cause the bending of a microcantilever (Fig. 2).10a,b,d A doubly bistable [3]rotaxane with four stations is first of all assembled such that the two rings of the [3]rotaxane are attached to the surface. Upon oxidation of the outer tetrathiafulvalene (TTF) stations, the cyclobis(paraquat-p-phenylene) ( CBPQT4+) ring shifts away from the outermost stations to the inner two 1,5-dioxynaphthalene (DNP) stations. This shuttling causes the rotaxane and thus the microcantilever beams to contract in a reversible and repeatable manner. In order to prove that the mechanism of the microcantilever deformation is in fact caused by the nanoscale motion of the switchable [3]rotaxane, a series of rigorous controls were investigated.10a Initial controls indicated that merely exposing the gold cantilevers to the chemical oxidant used to activate the molecular switch did not alone elicit any significant deformation. However, in order to rule out the effects of other forces that are created by the oxidation of the TTF moiety—such as electrostatic repulsion or interactions between the switching components and the surface—additional control experiments were carried out. By attaching the dumbbell portion of the [3]rotaxane to the surface of the gold cantilever, the effects of TTF oxidation can be measured. Upon oxidation of TTF to TTF2+, the cantilever has been observed to bend in the opposite direction compared to the direction of bending witnessed when using a fully functioning doubly bistable [3]rotaxane switch. This observation indicates that charge repulsion is dominant when one of the key components—e.g., the CBPQT4+ ring—for mechanical switching is not present and that the directional force created by the monolayer of nanoscale machines is able to overcome this repulsion, resulting in the microcantilever deforming in the expected direction.
![A thin gold surface coated with palindromic bistable [3]rotaxanes shows how chemical and electrochemical energy can be employed to bend a microcantilever, i.e., how chemical or electrochemical energy can be converted into mechanical energy. The tethered palindromic [3]rotaxane (TPR8+) contains two tetrathiafulvalene (TTF) stations near the termini of the dumbbell and two 1,5-dioxynaphthalene (DNP) stations at the center. The two electron deficient cyclobis(paraquat-p-phenylene) ( CBPQT4+) rings bear disulfide tethers such that TPR8+ can be assembled onto the gold surface. Oxidation of TTF by either chemical or electrochemical activation causes the shuttling of the CBPQT4+ rings to the DNP sites, shortening the inter-ring distance from 4.2 to 1.4 nm in a doubly bistable [3]rotaxane. The contraction of the microcantilever coated with TPR8+ has been achieved both chemically10b and electrochemically.10d](/image/article/2011/SC/c0sc00453g/c0sc00453g-f2.gif) | ||
| Fig. 2 A thin gold surface coated with palindromic bistable [3]rotaxanes shows how chemical and electrochemical energy can be employed to bend a microcantilever, i.e., how chemical or electrochemical energy can be converted into mechanical energy. The tethered palindromic [3]rotaxane (TPR8+) contains two tetrathiafulvalene (TTF) stations near the termini of the dumbbell and two 1,5-dioxynaphthalene (DNP) stations at the center. The two electron deficient cyclobis(paraquat-p-phenylene) ( CBPQT4+) rings bear disulfide tethers such that TPR8+ can be assembled onto the gold surface. Oxidation of TTF by either chemical or electrochemical activation causes the shuttling of the CBPQT4+ rings to the DNP sites, shortening the inter-ring distance from 4.2 to 1.4 nm in a doubly bistable [3]rotaxane. The contraction of the microcantilever coated with TPR8+ has been achieved both chemically10b and electrochemically.10d | ||
The mobility of a substance on a surface can also be influenced by coating the surface with responsive molecules, such as bistable rotaxanes or catenanes. A self-assembled monolayer (SAM) containing a bistable [2]rotaxane, for example, has been developed20 that is capable of moving a drop of liquid by irradiation with UV light (Fig. 3). The two stations of the rotaxane consist of a fluorinated region and a photo-switchable olefinic component. When the SAM is irradiated, some of the olefins are isomerised from (E) to (Z) configurations, causing the ring to shuttle onto the fluorinated region of the dumbell. This isomerisation results in a change in the hydrophobicity of the surface and thus the interaction between the surface and the droplet becomes very different. Using this actuation at the molecular level, a droplet of diiodomethane can be moved several millimetres and even up a 12° incline.
![A SAM of photo-switchable bistable [2]rotaxanes, capable of causing the movement of a droplet of diiodomethane along a surface.20 The bistable [2]rotaxanes are composed of a fumaramide station (green) and a tetrafluorosuccinamide station (orange). Under ambient conditions, the ring has a higher affinity for the fumaramide station. Irradiation with UV light isomerises the fumaramide into a conformation with less affinity for the ring, inducing it to shuttle to the fluorinated station. This movement causes an alteration in the hydrophobicity of the surface, resulting in a change in the contact angle between the droplet and the surface. This physical adjustment causes the droplet to spread and eventually move across the surface. Hence, irradiation of the SAM can result in the movement of a droplet of diiodomethane up an incline as large as 12°.](/image/article/2011/SC/c0sc00453g/c0sc00453g-f3.gif) | ||
| Fig. 3 A SAM of photo-switchable bistable [2]rotaxanes, capable of causing the movement of a droplet of diiodomethane along a surface.20 The bistable [2]rotaxanes are composed of a fumaramide station (green) and a tetrafluorosuccinamide station (orange). Under ambient conditions, the ring has a higher affinity for the fumaramide station. Irradiation with UV light isomerises the fumaramide into a conformation with less affinity for the ring, inducing it to shuttle to the fluorinated station. This movement causes an alteration in the hydrophobicity of the surface, resulting in a change in the contact angle between the droplet and the surface. This physical adjustment causes the droplet to spread and eventually move across the surface. Hence, irradiation of the SAM can result in the movement of a droplet of diiodomethane up an incline as large as 12°. | ||
A number of nanoparticle-based mechanised integrated systems have been fabricated recently10g–i during the course of a collaboration between ourselves and the Grzybowski group. These nanosystems display observable macroscopic properties which can be controlled by stimulating their molecular components. By attaching switchable molecules, which are known to participate in host–guest interactions to metal nanoparticles, one can instill a range of different properties into the integrated system. It has been demonstrated that guests, such as threads containing TTF and DNP units, can be appended to metal (gold, platinum, or palladium) nanoparticles and form pseudorotaxanes when exposed10g to hosts such as CBPQT4+. In the same investigation, we were also able to attach switchable catenanes to the surfaces of gold nanoparticles, and monitor the switching by changes in ζ potentials. More recently, gold nanoparticles were functionalised with a singleTTF guest molecule, such that they can then form10h nanoparticulate twins, triplets and quadruplets when exposed to dimers, trimers, or tetramers displaying two, three and four CBPQT4+ rings in turn. In a further extension of this work, nanoparticulate aggregates were assembled by using TTF-functionalised silver nanoparticles and DNP-functionalised gold nanoparticles in solution with polymer carrying pendant CBPQT4+ rings—namely poly(CBPQT4+).10i By altering the redox chemistry, the polymer's affinity for either the gold or silver nanoparticles can be controlled (Fig. 4). The polymer aggregates primarily with the silver nanoparticles in a reductive environment, thus freeing the gold nanoparticles into solution, such that the solution appears wine-coloured. Conversely, in an oxidative environment, the silver nanoparticles are responsible for the yellow colour of the solution when the gold nanoparticles are sequestered by the polymer.
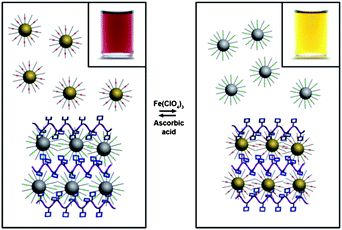 | ||
| Fig. 4 The switching of the solubility properties of functionalised gold and silver nanoparticles can be controlled using redox conditions. When an equimolar amount of TTF-functionalised silver nanoparticles and DNP-functionalised gold nanoparticles are exposed to poly(CBPQT4+), the silver nanoparticles (because of their higher affinity for CBPQT4+) aggregate with the polymer and precipitate, leaving the gold nanoparticles in solution (wine) as shown in the left panel.10i Upon adding Fe(ClO4)3, TTF is oxidised to TTF2+ and the silver nanoparticles are released into solution (yellow), allowing the DNP-functionalised gold nanoparticles to bind to poly(CBPQT4+) and precipitate out of solution (right panel). Adding ascorbic acid induces the reverse process, but with much slower kinetics. | ||
Not only can molecular scale motion be used to create macroscopic motion, it can also be used to mediate the controlled release of a cargo from a solid support, in turn effecting a macroscale response. A wide selection of (bistable) rotaxanes and pseudorotaxanes has been attached to the surfaces of mesoporous silica nanoparticles (MSNPs) in an attempt to control the flow of dyes and drugs into and out of the nanopores of the MSNPs for use in drug delivery.10c,e,f These nanoscopic devices function by controlling the translations of bulky rings along the stalks of the (pseudo)rotaxanes, a mechanical motion which orchestrates the opening and closing of the gates—depending on the conditions of the surrounding environment—positioned around the nanopores on the surface. These MSNPs can be controlled by redox chemistry,21 pH,22 light,23 and specific biological triggers.24
The proof-of-principle example10c of these “nanovalves” in action has involved the attachment of bistable rotaxanes to the surfaces of the MSNPs followed by showing that they can be opened and closed using redox chemistry (Fig. 5). Initially, the CBPQT4+ rings are stationed on TTF—well away from the nanopores. In this co-conformation, the gates are open, allowing the cargo to be loaded into the nanoparticle under a concentration gradient. On exercising oxidative conditions, the gates become closed as the CBPQT4+ rings station themselves on the DNP units. Subsequent reduction elicits the opening of the nanopores and the release of the cargo from the MSNPs. The functioning of this system, however, occurs in an organic solvent and requires a considerable effort in chemical synthesis. In a recent example,25 a water-soluble bistable [2]pseudorotaxane, assembled on the surfaces of MSNPs, employs (Fig. 6) cucurbit[6]uril (CB[6]) stationed on viologen units to block the nanopores. Upon lowering the pH, the CB[6] ring shuttles away from the nanopores, thus releasing the cargo. This water-soluble variant contains some of the key requirements for future applications in physiological environments.
![An early example of switchable bistable [2]rotaxanes attached to the surfaces of mesoporous silica nanoparticles (MSNPs).10c Initially, the bistable [2]rotaxanes are in a co-conformation such that the CBPQT4+ rings are stationed on TTF—far away from the nanopores. In this state, the gates are open, allowing diffusion of a cargo into the nanopores under a concentration gradient. Upon oxidation of TTF to TTF2+ with Fe(ClO4)3·6H2O, the bistable [2]rotaxanes adopt a co-conformation in which the CBPQT4+ rings encircle the DNP units, i.e., the gates are closed. Subsequently, the capped MSNPs can be opened by the addition of ascorbic acid to reduce TTF2+ back to TTF so that the bistable [2]rotaxane can reassume its ground state co-conformation, from whence the cargo can diffuse back out of the nanopores of the MSNPs. This system demonstrates controlled release of cargo in an organic solvent.](/image/article/2011/SC/c0sc00453g/c0sc00453g-f5.gif) | ||
| Fig. 5 An early example of switchable bistable [2]rotaxanes attached to the surfaces of mesoporous silica nanoparticles (MSNPs).10c Initially, the bistable [2]rotaxanes are in a co-conformation such that the CBPQT4+ rings are stationed on TTF—far away from the nanopores. In this state, the gates are open, allowing diffusion of a cargo into the nanopores under a concentration gradient. Upon oxidation of TTF to TTF2+ with Fe(ClO4)3·6H2O, the bistable [2]rotaxanes adopt a co-conformation in which the CBPQT4+ rings encircle the DNP units, i.e., the gates are closed. Subsequently, the capped MSNPs can be opened by the addition of ascorbic acid to reduce TTF2+ back to TTF so that the bistable [2]rotaxane can reassume its ground state co-conformation, from whence the cargo can diffuse back out of the nanopores of the MSNPs. This system demonstrates controlled release of cargo in an organic solvent. | ||
![Cucurbit[6]uril-(CB[6])-capped MSNPs exhibit cargo release at low pH.25 A linear stalk containing a viologen binding site and terminated by a carboxylic acid is appended to the surface of the MSNPs. Uncapped nanoparticles can be loaded and capped at neutral pH, before submitting them to release conditions. At pH 4 or less, CB[6] has a higher affinity for the protonated carboxylic acid than it has for the viologen units on the stalks, allowing for cargo molecules to traverse in and out of the nanopores freely. This system builds upon previous MSNP work with the added advantage that it functions in water.](/image/article/2011/SC/c0sc00453g/c0sc00453g-f6.gif) | ||
| Fig. 6 Cucurbit[6]uril-(CB[6])-capped MSNPs exhibit cargo release at low pH.25 A linear stalk containing a viologen binding site and terminated by a carboxylic acid is appended to the surface of the MSNPs. Uncapped nanoparticles can be loaded and capped at neutral pH, before submitting them to release conditions. At pH 4 or less, CB[6] has a higher affinity for the protonated carboxylic acid than it has for the viologen units on the stalks, allowing for cargo molecules to traverse in and out of the nanopores freely. This system builds upon previous MSNP work with the added advantage that it functions in water. | ||
The immobilisation of molecular machines in robust environments has revolutionised the way we apply mechanostereochemical principles to the fabrication of functioning devices. We have highlighted a few successes in the mobilisation of objects on surfaces and in the deformation of materials, as well as draw attention to potential applications in drug delivery. While these examples represent significant advances in the field, they are no more than tiny steps; there are still many challenges to be faced.
Bioconjugated molecular devices
Just as harnessing function and motion from artificial molecular machines requires utilising new platforms and materials, scientists and engineers can access new methods of translational motion by tethering biomolecules to synthetic surfaces. DNA, for example, is a naturally occurring biopolymer which is unique with respect to its highly programmable structure and synthetically accessible nature, making it an ideal material for the design and fabrication of biomolecular machines. The exploitation of the highly specific nature of DNA hybridisation permits the use of a series of chain-exchange reactions to drive nanomachines, as first demonstrated with the so-called DNA tweezers.26 Here, the addition of a substrate (alternating single-stranded DNA) triggers the opening and closing of a pair of nanotweezers. Similarly, DNA machines can be designed such that the addition of short oligonucleotide substrates elicits the unidirectional motion of a DNA “walker” along a track.27 Synthetic DNA systems may be used to effect macroscale movements in the form of contractions and expansions of biopolymers, powered by the addition of single-stranded DNA substrates.28 Immobilisation of such designs on a surface represents a major step in the progression towards functioning devices. Examples of this immobilisation include DNA tweezers that are localised along a track that template amine acylation coupling reactions29 and DNA nanomachines which can bend a microcantilever.30Biomolecules which act as molecular machines in their natural environment can be used to fabricate synthetic devices. These devices take advantage of both translational and rotational motion provided by Nature in order to effect macroscopic movements on a synthetic platform. One example utilises a hybrid nanodevice which is composed of three components: an engineered Ni post, an F1-ATPase biomolecular motor, and Ni nanopropellers appended to the motors.31 This bioconjugate nanodevice creates rotational motion that can be controlled by the addition of ATP to power the motor. Similarly, the gliding bacteria Mycoplasma mobile has been used to power a silicon dioxide rotor on a silicon track.32 The translational movement of the bacteria can be harnessed to display rotational speeds up to 2.6 rpm. Here, the merging of a living organism with inorganic materials results in a hybrid mechanical device.
Conclusion
When it comes to designing and making—by dint of synthesis and fabrication—complex integrated systems which perform functions on the nano-, micro- and macroscopic scales, chemists need to forsake solution for surfaces, interfaces and frameworks in order to develop the requisite platforms for the precise positioning in one-, two-, and three-dimensional space of switchable mechanically interlocked molecules and the appropriate mounting of molecular machinery. The act of transferring the concepts of molecular recognition and self-assembly from the biological world—often through the practice of templation in synthesis—into a chemical context can very often provide the stepping stone for the creation of integrated complex systems. In this mini-review, we have highlighted a few of the early successes in the realm of integrated systems where the responses of molecular switches and the movement of molecular machines are expressed on the nano-, micro- and macroscopic scales as a result of chemical (pH, redox), electrochemical (redox) and photochemical (configurational change, redox) stimuli. The introduction of molecular switches and machines into condensed phases on surfaces (SAMs), at molecular switch tunnel junctions (MSTJs) and inside frameworks (MOFs) is only reminiscent of living systems in the most cursory of manners. The need for chemists and materials scientists to wear their chemistry and science on their sleeves and sever the umbilical cord to the life sciences in terms of their desire to do no more than merely replicate its behaviour using simplistic chemical “mimics” is becoming more apparent as time progresses.Future directions
While working to design and construct unique integrated systems that draw inspiration from Nature but don't simply mimic it, we may still turn to biological media as platforms for the mounting of switchable machinery in order to enhance mechanostereochemistry33 and give expression to its potential consequences. Mechanical motion can be achieved through the conjugation of unnatural surfaces or compounds with biomolecules which include synthetic DNA, as well as tethered and modified proteins. Sophisticated devices and machines on the molecular scale can be synthesised, but now is the time to devise new, more complex, “mechanised” materials. The fundamental groundwork for effecting a merger of these two fields exists. We can readily functionalise biomolecules through the conjugation of amines34 and of thiols (cysteine residues),35 by site-directed mutagenesis,36 and through the marriage of azides and alkynes using Huisgen “click” chemistry,37 or by the incorporation of reactive groups using solid-phase synthesis. Similarly, we can rather easily alter regions of various biomolecular systems and machines by synthetic methods. Examples of these hybrid systems have been visited extensively in DNA nanotechnology. DNA has been functionalised with metal centers,38gold nanoparticles,39 switchable molecules,40 and hosts/guests41 in order to effect new architectures, properties and functions. Just as synthetic materials are being used as stents and joints in medicine, synthetic materials could be used as “molecular prosthetics,” giving us control over biological processes using correlated nanoscale motions. The works of Hoffman, Stayton and co-workers5a–c are early examples of this type of molecular prosthetics. Site-specific conjugation of various collapsible polymers to the active site domain of an enzyme acts as a door to the catalytic site. Similarly, a thermoresponsive polymer can be used (Fig. 7) to block the DNA recognition site of a multi-subunit DNA motor5d in a controllable fashion. Employing our knowledge of switchable molecules and applying them in different contexts—e.g., in biological media or at surfaces, interfaces and frameworks in fabricated materials—may allow us to empower artificial molecular machinery and unleash its latent potential. In the realms of device fabrication and the development of “smart” therapeutics, controlled responses are becoming increasingly the order of the day. It is imperative to build upon the achievements of chemists in the design and synthesis of artificial molecular switches and machines so that scientists and engineers may continue to introduce highly programmable molecular devices into robust environments in search of the next generation of mechanised materials.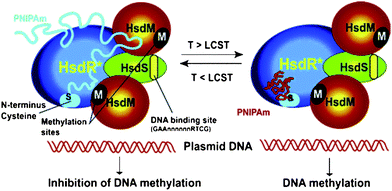 | ||
| Fig. 7 A thermoresponsive polymer conjugated to a subunit distal to the DNA binding site of a DNA motor protein in order to regulate function.5d A hybrid DNA restriction-modification (R-M) EcoR124I enzyme based on the natural type I R-M system is illustrated graphically. Native EcoR124I contains three types of subunits: one HsdS which is responsible for DNA sequence recognition; two HsdMs are responsible for DNA methylation; two HsdRs are responsible for DNA cleavage. Here, the hybrid enzyme has one sub-assembly missing one HsdR subunit such that it exhibits methylase activity but translocates DNA without restriction (cleavage). At temperatures below the lower critical solution temperature (LCST ∼32 °C), the thermoresponsive polymer poly(N-isopropylacrylamide) (PNIPAm) is capable of blocking the DNA recognition site as it is soluble and expanded around the binding site. The polymer contracts and becomes insoluble at temperatures higher than the LCST to expose the site for DNA methylation. Initial studies have shown that conjugation of the polymer at the methylation sites irreversibly blocks enzyme activity. However, conjugation distal to the methylation site reveals approximately 10% reaction at temperatures lower than the LCST and approximately 90–100% methylation at temperatures higher than LCST. Hence, low temperatures inhibit DNA methylation and high temperatures enable it. | ||
Acknowledgements
This work was sponsored by the National Center for Nano Technology Research at the King Abdulaziz City for Science and Technology (KACST) in Saudi Arabia. The authors thank Dr Turki S. Al-Saud and Dr Soliman H. Alkhowaiter at KACST for their generous support of this program of research at Northwestern University.Notes and references
- I. Rayment, H. M. Holden, M. Whittaker, C. B. Yohn, M. Lorenz, K. C. Holmes and R. A. Milligan, Science, 1993, 261, 58–65 CrossRef CAS.
- F. J. Kull, E. P. Sablin, R. Lau, R. J. Fletterick and R. D. Vale, Nature, 1996, 380, 550–555 CrossRef.
- J. P. Abrahams, A. G. W. Leslie, R. Lutter and J. E. Walker, Nature, 1994, 370, 621–628 CrossRef CAS.
- J. S. Berg, B. C. Powell and R. E. Cheney, Mol. Biol. Cell, 2001, 12, 780–794 CAS.
- (a) A. Chilkoti, G. H. Chen, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1994, 5, 504–507 CrossRef CAS; (b) T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS; (c) T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517–525 CrossRef CAS; (d) S. S. Pennadam, M. D. Lavigne, C. F. Dutta, K. Firman, D. Mernagh, D. C. Górecki and C. Alexander, J. Am. Chem. Soc., 2004, 126, 13208–13209 CrossRef CAS.
- (a) H. Hess, J. Clemmens, D. Qin, J. Howard and V. Vogel, Nano Lett., 2001, 1, 235–239 CrossRef CAS; (b) H. Liu, J. J. Schmidt, G. D. Bachand, S. S. Rizk, L. L. Looger, H. W. Hellinga and C. D. Montemagno, Nature Mater., 2002, 1, 173–177 CrossRef CAS; (c) S. Diez, C. Reuther, C. Dinu, R. Siedel, M. Mertig, W. Pompe and J. Howard, Nano Lett., 2003, 3, 1251–1254 CrossRef CAS; (d) H. Hess, G. D. Bachand and V. Vogel, Chem.–Eur. J., 2004, 10, 2110–2116 CrossRef CAS; (e) M. G. L. Van den Heuvel and C. Dekker, Science, 2007, 317, 333–336 CrossRef; (f) W. Song, Q. He, Y. Ciu, H. Mohwald, S. Diez and J. Li, Biochem. Biophys. Res. Commun., 2009, 379, 175–178 CrossRef CAS; (g) H. Takatsuki, K. M. Rice, S. Asano, B. S. Day, M. Hino, K. Oiwa, R. Ishikawa, Y. Hiratsuka, T. Q. P. Uyeda, K. Kohamaand and E. R. Blough, Small, 2010, 6, 452–457 CrossRef CAS.
- (a) V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS; (b) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem. Int. Ed, 2000, 39, 3348–3391 CrossRef CAS; (c) W. R. Browne and B. L. Feringa, Nature Nanotechnol., 2006, 1, 25–35 CrossRef CAS; (d) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2006, 46, 72–191; (e) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH Verlag, 2008 Search PubMed.
- (a) A. M. Schoevaars, W. Kruzinga, R. W. J. Zijlstra, N. Veldman, A. L. Spek and B. L. Feringa, J. Org. Chem., 1997, 62, 4943–4948 CrossRef CAS; (b) T. R. Kelly, H. De Sliva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS; (c) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS; (d) T. R. Kelly, X. Cai, F. Damkaci, S. B. Panicker, B. Tu, S. M. Bushell, I. Cornella, M. J. Piggott, R. Salives, M. Cavero, Y. Zhao and S. Jasmin, J. Am. Chem. Soc., 2006, 129, 376–380; (e) A. A. Kulago, E. M. Mes, M. Klok, A. Meetsma, A. M. Brouwer and B. L. Feringa, J. Org. Chem., 2010, 75, 666–679 CrossRef CAS.
- (a) P.-L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef; (b) R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef; (c) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS; (d) C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS; (e) Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H.-R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS; (f) J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. Delonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS.
- (a) T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS; (b) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS; (c) T. D. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS; (d) B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y.-W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291–300 CrossRef CAS; (e) S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 12912–12914 CrossRef CAS; (f) D. Ferris, Y.-L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS; (g) R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233–4235 CrossRef CAS; (h) M. A. Olson, A. Coskun, R. Klajn, L. Fang, S. K. Dey, K. P. Browne, B. A. Grzybowski and J. F. Stoddart, Nano Lett., 2009, 9, 3185–3190 CrossRef CAS; (i) R. Klajn, M. A. Olson, P. J. Wesson, L. Fang, A. Coskun, A. Trabolsi, S. Soh, J. F. Stoddart and B. A. Grzybowski, Nature Chem., 2009, 1, 733–738 CrossRef CAS; (j) T. Ye, A. S. Kumar, S. Saha, T. Takami, T. J. Huang, J. F. Stoddart and P. S. Weiss, ACS Nano, 2010, 4, 3697–3701 CrossRef CAS.
- (a) C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632–12641 CrossRef CAS; (b) R. Beckman, K. Beverly, A. Boukai, Y. Bunimovich, J. W. Choi, E. DeIonno, J. Green, E. Johnston-Halperin, Y. Luo, B. Sheriff, J. F. Stoddart and J. R. Heath, Faraday Discuss., 2006, 131, 9–22 RSC; (c) J. R. Heath, Annu. Rev. Mater. Res., 2009, 39, 1–23 CrossRef CAS; (d) S. J. Van der Molen and P. Liljeroth, J. Phys.: Condens. Matter, 2010, 22, 1–30.
- (a) J. F. Stoddart and H. M. Colquhoun, Tetrahedron, 2008, 64, 8231–8236 CrossRef CAS; (b) J. F. Stoddart, Nature Chem., 2009, 1, 14–15 CrossRef CAS; (c) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; (d) R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC; (e) H. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nature Chem., 2010, 2, 439–443 CrossRef CAS.
- (a) J.-F. Nierengarten, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375–376 CrossRef CAS; (b) M.-V. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1997, 36, 1904–1907 CrossRef CAS; (c) V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924–1936 CrossRef CAS; (d) J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Angew. Chem., Int. Ed., 2001, 40, 1216–1221 CrossRef CAS; (e) Y.-L. Zhao, W. R. Dichtel, A. Trabolsi, S. Saha, I. Aprahamian and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 11294–11296 CrossRef CAS.
- (a) O. M. Yaghi, G. Li and H. Li, Nature, 1995, 117, 10401–10402 CAS; (b) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS; (c) M. Eddaoudi, D. Moler, H. Li, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS; (d) N. L. Rosi, M. Eddaoudi, D. T. Vodak, J. Eckert, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS; (e) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS; (f) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS; (g) C. Volkringer, D. Popov, T. Loiseau, N. Guillou, G. Férey, M. Haouas, F. Taulelle, C. Mellot-Draznieks, M. Burghammer and C. Riekel, Nature Mater., 2007, 6, 760–764 CrossRef CAS; (h) T. Uemura, N. Yanai and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228 RSC; (i) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nature Mater., 2010, 9, 172–178 CrossRef CAS; (j) H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS.
- (a) Q. Li, W. Zhang, O.Š. Miljanić, C.-H. Sue, Y.-L. Zhao, L. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS; (b) Y.-L. Zhao, L. Liu, W. Zhang, C.-H. Sue, Q. Li, O.Š. Miljanić, O. M. Yaghi and J. F. Stoddart, Chem. Eur. J., 2009, 15, 13356–13308; (c) Q. Li, W. Zhang, O.Š. Miljanić, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Chem. Commun., 2010, 46, 380–382 RSC.
- (a) M. Klok, W. R. Browne and B. L. Feringa, Phys. Chem. Chem. Phys., 2009, 11, 9124–9131 RSC; (b) E. M. Geertsema, S. J. van der Molen, M. Martens and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16919–16924 CrossRef CAS.
- (a) M. Ikeda, M. Takeuchi, S. Shinkai, F. Tani and Y. Naruta, Bull. Chem. Soc. Jpn., 2001, 74, 739–746 CrossRef; (b) X. Zheng, M. E. Mulcahy, D. Horinek, F. Galeotti, T. F. Magnera and J. Michl, J. Am. Chem. Soc., 2004, 126, 4540–4542 CrossRef CAS; (c) T. F. Magnera and J. Michl, Top. Curr. Chem., 2005, 262, 63–97 CAS; (d) J. Otsuki, S. Kawaguchi, T. Yamakawa, M. Asakawa and K. Miyake, Langmuir, 2006, 22, 5708–5715 CrossRef CAS.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nature Mater., 2005, 4, 704–710 CrossRef CAS.
- (a) R. Hernandez, H.-.R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS; (b) T. D. Nguyen, Y. Liu, S. Saha, K. C.-F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS.
- (a) T. D. Nguyen, K. C.-F. Leung, M. Liong, C. D. Pentecost, J. F. Stoddart and J. I. Zink, Org. Lett., 2006, 8, 3363–3366 CrossRef CAS; (b) K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Chem. Mater., 2006, 18, 5919–5928 CrossRef CAS.
- (a) N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS; (b) T. D. Nguyen, K. C.-F. Leung, M. Liong, Y. Liu, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2101–2110 CrossRef CAS; (c) K. K. Cotí, M. E. Belowich, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- (a) K. Patel, S. Angelos, W. R. Dichtel, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS; (b) M. W. Ambrogio, T. A. Percorelli, K. Patel, N. M. Khashab, A. Trabolsi, H. A. Khatib, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Org. Lett., 2010, 12, 3304–3307 CrossRef CAS.
- N. M. Khashab, M. E. Belowich, A. Trabolsi, D. C. Friedman, C. Valente, Y. Lau, H. A. Khatib, J. I. Zink and J. F. Stoddart, Chem. Commun., 2009, 5371–5373 RSC.
- B. Yurke, A. J. Turberfield, A. P. Mills Jr., F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS.
- (a) T. H. LaBean, H. Yan, J. Kopatsch, F. R. Liu, E. Winfree, J. H. Reif and N. C. Seeman, J. Am. Chem. Soc., 2000, 122, 1848–1860 CrossRef CAS; (b) J. S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS.
- D. Lubrich, J. Lin and J. Yan, Angew. Chem., Int. Ed., 2008, 47, 7026–7028 CrossRef CAS.
- R. Chhabra, J. Sharma, Y. Liu and H. Yan, Nano Lett., 2006, 6, 978–983 CrossRef CAS.
- W. Shu, D. Liu, M. W. C. K. Reiner, T. Strunz, M. E. Welland, S. Balsubramanian and R. A. McKendry, J. Am. Chem. Soc., 2005, 127, 17054–17060 CrossRef CAS.
- (a) R. K. Soong, G. D. Bachand, H. P. Neves, A. G. Olkhovetes, H. G. Craighead and C. D. Montemagno, Science, 2000, 290, 1555–1558 CrossRef CAS; (b) R. K. Soong, H. P. Neeves, J. J. Schmidt and C. D. Montemagno, Biomed. Microdevices, 2001, 3, 71–73 CrossRef CAS.
- Y. Hiratsuka, M. Miyata, T. Tada and T. Q. P. Uyeda, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13618–13623 CrossRef CAS.
- M. A. Olson, Y. Y. Botros and J. F. Stoddart, Pure Appl. Chem., 2010, 82, 1569–1574 CrossRef CAS.
- (a) T. Miron and M. Wilchek, Bioconjugate Chem., 1993, 4, 568–569 CrossRef CAS; (b) G. E. Francis, D. Fisher, C. Delgado, F. Malik, A. Gardiner and D. Neale, Int. J. Hematol., 1998, 68, 1–18 CrossRef CAS.
- (a) C. T. Kuan, Q. C. Wang and I. Pastan, J. Biol. Chem., 1994, 269, 7610–7616 CAS; (b) M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Bioconjugate Chem., 2001, 12, 1051–1056 CrossRef CAS; (c) A. S. M. Kamruzzahan, A. Ebner, L. Wildling, F. Kienberger, C. K. Riener, C. D. Hahn, P. D. Pollheimer, P. Winklehner, M. Hölzl, B. Lackner, D. M. Schörkl, P. Hinterdorfer and H. J. Gruber, Bioconjugate Chem., 2006, 17, 1473–1481 CrossRef CAS.
- Y. Yamamoto, Y. Tsutsumi, Y. Yoshioka, T. Nishibata, K. Kobayashi, T. Okamoto, Y. Mukai, T. Shimizu, S. Nakagawa, S. Nagata and T. Mayumi, Nat. Biotechnol., 2003, 21, 546–552 CrossRef CAS.
- (a) R. Hüisgen, Pure Appl. Chem., 1989, 61, 613–628 CrossRef CAS; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (c) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- (a) M. Göritz and R. Krämer, J. Am. Chem. Soc., 2005, 127, 18016–18017 CrossRef; (b) H. Yang and H. Sleiman, Angew. Chem., Int. Ed., 2008, 47, 2443–2446 CrossRef CAS; (c) H. Yang, A. Z. Rys, C. K. McLaughlin and H. Sleiman, Angew. Chem., Int. Ed., 2009, 48, 9919–9923 CrossRef CAS.
- Y. Wen, C. K. McLaughlin, P. K. Lo, H. Yang and H. Sleiman, Bioconjugate Chem., 2010, 21, 1413–1416 CrossRef CAS.
- M. Zhou, X. Liang, T. Mochizuki and H. Asanuma, Angew. Chem., Int. Ed., 2010, 49, 2167–2170 CrossRef CAS.
- (a) T. Ihara, A. Uemura, A. Futamura, M. Shimizu, N. Baba, S. Nishizawa, N. Teramae and A. Jyo, J. Am. Chem. Soc., 2009, 131, 1386–1387 CrossRef CAS; (b) A. Kuzuya, T. Ohnishi, T. Wasano, S. Nagaoka, J. Sumaoka, T. Ihara, A. Jyo and M. Komiyama, Bioconjugate Chem., 2009, 20, 1643–1649 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |