Genetically encoding an aliphatic diazirine for protein photocrosslinking†
Chungjung
Chou
a,
Rajendra
Uprety
a,
Lloyd
Davis
b,
Jason W.
Chin
*b and
Alexander
Deiters
*a
aNorth Carolina State University, Department of Chemistry, Raleigh, NC 27695, USA. E-mail: alex_deiters@ncsu.edu
bMedical Research Council Laboratory of Molecular Biology, Hills Rd, Cambridge, CB2 0QH, UK. E-mail: chin@mrc-lmb.cam.ac.uk
First published on 1st December 2010
Abstract
Photocrosslinking is an important approach that allows discovery and detailed investigation of protein–protein, protein–oligonucleotide, and protein–small molecule interactions with high temporal and spatial resolution. A major limitation to the universal application of this methodology is the site-specific introduction of efficient aliphatic photocrosslinking probes into proteins of interest. Here, we report a novel aliphatic diazirine amino acid and its genetically encoded, site-specific incorporation into proteins in bacterial and mammalian cells. Furthermore, we demonstrate efficient photocrosslinking of a test proteinin vitro and in vivo.
Introduction
Photocrosslinking of biological molecules is a powerful method for direct structure–function studies of physiological processes, since it can provide time-resolved information on the spatial relationship of molecular components in a biological system.1,2 The central element of a photocrosslinking approach is a photoaffinity label, that, when activated by UV light of a suitable wavelength, forms a highly reactive species, which is available for the formation of a covalent bond with regions of a biomolecule in van der Waals contact. Photocrosslinking approaches have been successfully applied in the mapping of protein–lipid interactions,3 small molecule–protein interactions,4oligonucleotide interactions,5oligonucleotide–protein,6 and, importantly, protein–protein interactions.7 Together with site-directed mutagenesis studies, NMR spectroscopy, and X-ray crystallography, photocrosslinking methods provide important tools for the identification of binding partners, as well as the mapping of binding sites and active sites.8A wide range of photocrosslinking groups have been developed with utility in biological studies, including azides, diazirines, and benzophenones.1,2 These photocrosslinking groups fit some or most of the following criteria: a) the group must be inert until UV irradiation, b) photoactivation should not damage the biological system under study, c) the activated group should undergo covalent bond formation with any biological functionality in close proximity, e.g., CH bonds, as well as more acidic NH and OH bonds, d) the photocrosslinking group should not substantially perturb the biological function of the investigated molecule, and e) the site-specific installation of the group should be easy.
The installation of photocrosslinking labels into biological molecules remains a major challenge and often requires substantial synthetic effort. The incorporation of a photoaffinity probe at a defined site in proteins is particularly challenging and has spurred substantial work, including post-translational bioconjugation,9 total synthesis of peptides,2,10in vitrotranscription/translation using misacylated tRNAs,11 and in vivo unnatural amino acid mutagenesis using an expanded genetic code.12–14 The latter methodology has the advantage that a) photoaffinity probes can be incorporated site-specifically into any protein, b) only standard molecular and cell biology techniques are required, and c) the protein containing the photocrosslinker can be expressed intracellularly in its native environment allowing for the direct investigation of native protein complexes. This may enable the isolation of weak or transient interactions that are not captured by non-covalent affinity purification methods such as tandem affinity purification (TAP) tagging that are commonly used in proteomic approaches.15
Phenylalanine derivatives containing azido, benzophenone, and diazirine groups have been site-specifically incorporated into proteins in E. coli in response to the amber codon using engineered variants of the M. jannaschiityrosyl-tRNA synthetase/tRNACUA pair.12,14para-Benzoyl-L-phenylalanine has also been incorporated site-specifically into proteins in yeast and mammalian cells using an E. colityrosyl-tRNA synthetase variant that was evolved in yeast to recognize this unnatural amino acid.16 The site-specific incorporation of benzophenone has been used to investigate and define protein interactions in a wide range of biological systems, especially protein interactions in large molecular machines or in biological membranes where many other methods for defining molecular interactions are challenging to employ.17 Despite the demonstrated utility of this approach in providing unique biological insight, the benzophenone group is very sterically demanding, and when placed at a protein–protein interface may destroy the very protein interaction under investigation. Indeed, since aromatic amino acids often constitute protein hotspots on protein surfaces,18 conservative substitutions of aromatic amino acids with benzophenone may often hamper the binding event under investigation. Non-conservative substitutions of the polar and charged residues, typically found on the surface of proteins, with benzophenone may interfere with the protein's solubility or folding. Hence, a pressing need exists for new genetically encoded, aliphatic, crosslinking amino acids that can be used to complement the use of p-benzoyl-L-phenylalanine and other aromatic photocrosslinking amino acids in investigating protein interactions.
Results & discussion
We discovered that pyrrolysyl tRNA synthetase/tRNACUA pairs can be used to introduce the lysine-based diazirine amino acid 1 into proteins in E. coli and in mammalian cells in response to the amber codon, TAG, with good yield. Furthermore, we demonstrate that the genetically encoded amino acid can be used to specifically photocrosslink protein–protein interactions (Scheme 1A).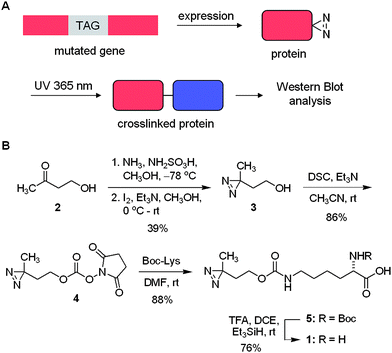 | ||
| Scheme 1 A) Unnatural amino acid mutagenesis in bacterial and mammalian cells allows for the site-specific incorporation of the diazirine group into proteins in response to an amber codon, TAG. Photochemical crosslinking then enables the identification of protein binding partners by analytical methods including Western blot and mass spec. B) Synthesis of the diazirine-modified lysine 1. DSC = disuccinimide carbonate, Boc = tert-butyloxycarbonyl, TFA = trifluoroacteic acid, DCE = dichloroethane. | ||
First, we synthesized the diazirine-lysine 1 starting with commercially available 4-hydroxybutan-2-one (2) and converting it in two steps into the corresponding diazirine 3 (Scheme 1B). Subsequent activation with disuccinimide carbonate (DSC) in the presence of triethylamine yields 4 in 86% after purification. Reaction of 4 with lysine bearing a Boc-protected α-amino group proceeded smoothly, delivering 5 in 88% yield. Removal of the Boc group proved to be more difficult than anticipated due to the acid sensitive nature of the diazirine group. Several common protocols (e.g., TFA/DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 or 1:1, at 0 °C or rt); HCl in methanol (1.0 or 0.5 M, at 0 °C or rt); HCl in dioxane/DCM (1.0 M, at 0 °C or rt); all with or without Me2S as a cation scavenger) were employed and reaction progress was monitored at different time intervals. All of the common conditions led to the decomposition of the starting material; however, clean deprotection was eventually achieved with a 1:20 mixture of TFA/dichloroethane in the presence of triethylsilane.19 The pure amino acid 1 was obtained in 76% yield after recrystallization.
:4 or 1:1, at 0 °C or rt); HCl in methanol (1.0 or 0.5 M, at 0 °C or rt); HCl in dioxane/DCM (1.0 M, at 0 °C or rt); all with or without Me2S as a cation scavenger) were employed and reaction progress was monitored at different time intervals. All of the common conditions led to the decomposition of the starting material; however, clean deprotection was eventually achieved with a 1:20 mixture of TFA/dichloroethane in the presence of triethylsilane.19 The pure amino acid 1 was obtained in 76% yield after recrystallization.
Previously, we20,21 and others22,23 demonstrated that the pyrrolysyl tRNA/tRNA synthetase pairs (PylRS/PyltRNACUA) from M. barkerii and M. mazei are orthogonal to all endogenous amino acids and all endogenous tRNAs and tRNA synthetases in E. coli and mammalian cells. Moreover, the wild-type synthetase PylRS is able to accommodate a wide range of non-natural lysine derivatives, generated from the modification of the ε-amino group with a variety of different groups, including tert-butyl, azido, and alkynyl24,25 linked via a carbamate to the ε-amino group. In addition, we demonstrated that PylRS can be evolved, through directed evolution approaches, to accept additional substrates, including the important post-translational modification acetyl-lysine21 and a sterically demanding photocaged lysine as substrates.26
To investigate whether 1 is a substrate for the PylRS/PyltRNACUA pair we transformed E. coli Top10 competent cells with pMyo4TAGpylT,21 containing a hexahistidine-tagged myoglobin gene with an amber stop codon, TAG, in the 4 position and pBKpylS, which contains the gene encoding MbPylRS.21Protein expression was conducted in 2YT media containing 1 (1 mM) and arabinose (0.04% after induction) at room temperature overnight and protein was purified by Ni-NTA chromatography. A yield of ∼2 mg myoglobin-1 per L of bacterial culture was obtained, which is comparable to the incorporation of other amino acids with the pyrrolysyl-tRNA synthetase/tRNA pair.25 The Coomassie stained SDS PAGE of the purified protein (Fig. 1A) reveals a clean 18 kDa band for myoglobin from the expression in the presence of 1, while no such band was found in the expression in the absence of 1. The purified protein was analyzed by ESI-MS and the deconvoluted mass spectrum revealed a molecular weight of 18521.94 ± 0.50 Da, which closely matches the expected value of 18522.26 for myoglobin containing 1 at the 4 position (Fig. 1B). Taken together these data demonstrate that 1 is an efficient substrate for the PylRS/PyltRNACUA pair.
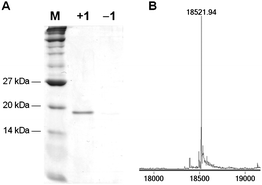 | ||
| Fig. 1 A) Coomassie stained gel of Ni-NTA purified myoglobin-4TAG expression in the presence (+1) and the absence (−1) of the diazirine-lysine. B) Incorporation of 1 into myoglobin was confirmed by ESI-MS, only showing a peak at 18521.94 Da in agreement with myo-1 (expected mass of 18522.26 Da). Myoglobin containing a natural amino acid at position 4 was not detected (e.g., lysine, 18396.15 Da). | ||
Since the pyrrolysyl-tRNA synthetase/tRNA pairs are orthogonal in both E. coli and mammalian cells21,23 we investigated the incorporation of 1 into proteins in human embryonic kidney (HEK293) cells in response to the amber stop codon. We examined the red and green fluorescence of HEK293 cells co-transfected with two constructs: a reporter construct (Fig. 2A)26 bearing an N-terminal mCherry gene, a linker containing an amber stop codon (TAG), a C-terminal enhanced GFP gene, a HA-tag coding sequence, and a PylRS/PyltRNACUA expression plasmid. As expected, mCherry fluorescence was detected from transfected HEK293 cells in the presence and absence of 1, but EGFP fluorescence was observed only upon addition of 1 (1 mM) to the cell culture media (Fig. 2B). Similarly HA-tagged mCherry-eGFP-HA fusion was detected with an anti-HA antibody only in the presence of 1 (Fig. 2C). These experiments demonstrate the efficient incorporation of 1 in response to the amber codon, TAG, in mCherry-TAG-eGFP-HA in mammalian cells.
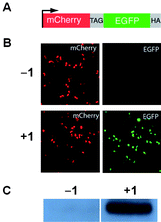 | ||
| Fig. 2 A) The mCherry-TAG-EGFP-HA reporter construct. B)Fluorescence micrographs of HEK293 cells expressing mCherry-TAG-EGFP-HA, PylRS, and PyltRNACUA in the absence and presence of 1. Full-length fusion protein is only generated in the presence of 1. C) Western blot of HEK293 cells expressing mCherry-TAG-EGFP-HA with an anti-HA tag antibody. | ||
To begin to demonstrate the utility of genetically encoding the diazirine 1 for the photocrosslinking of protein–protein interactions, we incorporated the diazirine into position 52 of glutathione S-transferase (GST), which is at the interface between monomers in the GST dimer.27 pGST52TAGpylT and pBKpylS were transformed into Top10 E. colicells and expressed as previously described for myoglobin.12,13 The hexahistidine-tagged GST protein was purified by Ni-NTA chromatography and photocrosslinked for 20 min through irradiation with UV light of 365 nm (transilluminator, 25 W). The protein was then analyzed by Western blot using an anti-His-tag antibody (Fig. 3A). His-tagged protein bands for the GST dimer were detected when the samples were exposed to UV light at 365 nm and when the protein was expressed in the presence of 1. We observed trace levels of protein expression in the absence of 1, as has previously been reported for unnatural amino acid incorporation experiments in rich media. Mass spectrometry experiments have shown that natural amino acids do not effectively compete for incorporation into the protein in response to the amber codon when unnatural amino acids are present.28
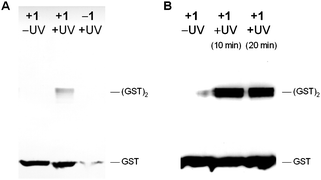 | ||
| Fig. 3 A) Immunoblot of photocrosslinked glutathione S-transferase (GST). GST-52TAG protein was expressed in the presence (+1) and the absence (−1) of the diazirine-lysine, purified, and either not irradiated (–UV) or irradiated (+UV) with UV light (365 nm, 20 min). The samples were analyzed by Western blot (8% SDS PAGE followed by detection with an anti-His antibody and 4CN colorimetric development). Photocrosslinked GST dimer was visible in the presence of both 1 and UV irradiation. B) Same as A), but photocrosslinking was conducted intracellularly and UV irradiation was performed for 10 and 20 min. | ||
To extend our approach to photocrosslinking in live bacterial cells, E. coli cultures producing GST-1 were washed, resuspended in PBS, and either kept in the dark or irradiated for 10–20 min (365 nm, 25 W, transilluminator). The cellular lysate was analyzed by Western blot, demonstrating efficient intracellular photocrosslinking and GST-dimer formation (Fig. 3B). Concentration effects may contribute to the better efficiency of crosslinking inside the cells compared to the in vitro experiment. These results indicate that the diazirine 1 can be efficiently incorporated into GST without interfering with natural protein dimer formation, and photochemical protein crosslinking could be induced via illumination of either purified GST containing 1 or intracellularly before protein isolation.
Conclusions
In summary, we have demonstrated that the pyrrolysyl-tRNA synthetase/tRNACUA pair can be used to incorporate a new aliphatic crosslinking amino acid site-specifically into proteins in E. coli and in mammalian cells. Moreover we have demonstrated the utility of the amino acid for photocrosslinking in vitro and in vivo. In future work we plan to extend our approach to studying biological processes in bacterial, mammalian, and yeast cells.29We acknowledge support by North Carolina State University and the Medical Research Council Laboratory of Molecular Biology. AD is a Beckman Young Investigator and a Cottrell Scholar. We thank Dr. T. A. Cropp (UMD) for the GST expression plasmid.
Notes and references
- J. Brunner, Annu. Rev. Biochem., 1993, 62, 483–514 CrossRef CAS; F. Kotzybahibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef; G. Dorman and G. D. Prestwich, Trends Biotechnol., 2000, 18, 64–77 CrossRef CAS; Y. Hatanaka and Y. Sadakane, Curr. Top. Med. Chem., 2002, 2, 271–288 CrossRef CAS; F. Knoll, T. Kolter and K. Sandhoff, Methods Enzymol., 2000, 311, 568–600 CAS.
- G. Dorman and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef CAS.
- J. Gubbens, E. Ruijter, L. E. V. de Fays, J. M. A. Damen, B. de Kruijff, M. Slijper, D. T. S. Rijkers, R. M. J. Liskamp and A. de Kroon, Chem. Biol., 2009, 16, 3–14 CrossRef CAS.
- A. L. MacKinnon, J. L. Garrison, R. S. Hegde and J. Taunton, J. Am. Chem. Soc., 2007, 129, 14560–14561 CrossRef CAS; M. R. Bond, H. C. Zhang, P. D. Vu and J. J. Kohler, Nat. Protoc., 2009, 4, 1044–1063 Search PubMed.
- O. Lentzen, J. F. Constant, E. Defrancq, M. Prevost, S. Schumm, C. Moucheron, P. Dumy and A. Kirsch-De Mesmaeker, ChemBioChem, 2003, 4, 195–202 CrossRef CAS.
- H. Geyer, R. Geyer and V. Pingoud, Nucleic Acids Res., 2004, 32 Search PubMed.
- M. Suchanek, A. Radzikowska and C. Thiele, Nat. Methods, 2005, 2, 261–267 CrossRef CAS.
- N. Hino, Y. Okazaki, T. Kobayashi, A. Hayashi, K. Sakamoto and S. Yokoyama, Nat. Methods, 2005, 2, 201–206 CrossRef CAS; R. Ieva and H. D. Bernstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19120–19125 CrossRef CAS.
- G. T. Hermanson, Academic Press, San Diego, Editon edn, 1996, pp. 528–569.
- J. C. Kauer, S. Ericksonviitanen, H. R. Wolfe and W. F. Degrado, J. Biol. Chem., 1986, 261, 695–700.
- T. Kanamori, S. Nishikawa, I. Shin, P. G. Schultz and T. Endo, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 485–490 CrossRef CAS.
- J. W. Chin, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11020–11024 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS; J. W. Chin and P. G. Schultz, ChemBioChem, 2002, 3, 1135–1137 CrossRef CAS.
- E. M. Tippmann, W. Liu, D. Summerer, A. V. Mack and P. G. Schultz, ChemBioChem, 2007, 8, 2210–2214 CrossRef.
- G. Rigaut, A. Shevchenko, B. Rutz, M. Wilm, M. Mann and B. Seraphin, Nat. Biotechnol., 1999, 17, 1030–1032 CrossRef CAS.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS.
- C. Liu, A. L. Young, A. Starling-Windhof, A. Bracher, S. Saschenbrecker, B. V. Rao, K. V. Rao, O. Berninghausen, T. Mielke, F. U. Hartl, R. Beckmann and M. Hayer-Hartl, Nature, 2010, 463, 197–202 CrossRef CAS; D. Braig, C. Bar, J. O. Thumfart and H. G. Koch, J. Mol. Biol., 2009, 390, 401–413 CrossRef CAS; S. Okuda and H. Tokuda, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5877–5882 CrossRef CAS; T. Haslberger, J. Weibezahn, R. Zahn, S. Lee, F. T. Tsai, B. Bukau and A. Mogk, Mol. Cell, 2007, 25, 247–260 CrossRef CAS; H. T. Chen, L. Warfield and S. Hahn, Nat. Struct. Mol. Biol., 2007, 14, 696–703 CrossRef CAS; H. Mori and K. Ito, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16159–16164 CrossRef CAS; C. M. Kaiser, H. C. Chang, V. R. Agashe, S. K. Lakshmipathy, S. A. Etchells, M. Hayer-Hartl, F. U. Hartl and J. M. Barral, Nature, 2006, 444, 455–460 CrossRef CAS; J. Weibezahn, P. Tessarz, C. Schlieker, R. Zahn, Z. Maglica, S. Lee, H. Zentgraf, E. U. Weber-Ban, D. A. Dougan, F. T. Tsai, A. Mogk and B. Bukau, Cell, 2004, 119, 653–665 CrossRef CAS.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- A. Mehta, R. Jaouhari, T. J. Benson and K. T. Douglas, Tetrahedron Lett, 1992, 33, 5441–5444 CrossRef CAS.
- H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441–444 CrossRef CAS.
- H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Chem. Biol., 2008, 4, 232–234 CrossRef CAS.
- W. Wan, Y. Huang, Z. Wang, W. K. Russell, P. J. Pai, D. H. Russell and W. R. Liu, Angew. Chem., Int. Ed., 2010, 49, 3211–3214 CrossRef CAS; T. Mukai, T. Kobayashi, N. Hino, T. Yanagisawa, K. Sakamoto and S. Yokoyama, Biochem. Biophys. Res. Commun., 2008, 371, 818–822 CrossRef CAS.
- Y. Huang, W. K. Russell, W. Wan, P. J. Pai, D. H. Russell and W. Liu, Mol. BioSyst., 2010, 6, 683–686 RSC; P. R. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. H. Geierstanger and P. G. Schultz, Angew. Chem., Int. Ed., 2009, 48, 4052–4055 CrossRef CAS; T. Yanagisawa, R. Ishii, R. Fukunaga, T. Kobayashi, K. Sakamoto and S. Yokoyama, Chem. Biol., 2008, 15, 1187–1197 CrossRef CAS.
- D. P. Nguyen, H. Lusic, H. Neumann, P. B. Kapadnis, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 8720 CrossRef CAS.
- D. P. Nguyen, M. M. Garcia Alai, P. B. Kapadnis, H. Neumann and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 14194–14195 CrossRef CAS.
- A. Gautier, D. P. Nguyen, H. Lusic, W. An, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 4086–4088 CrossRef CAS.
- K. Lim, J. X. Ho, K. Keeling, G. L. Gilliland, X. Ji, F. Ruker and D. C. Carter, Protein Sci., 1994, 3, 2233–2244 CrossRef CAS.
- K. Wang, H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Biotechnol., 2007, 25, 770–777 CrossRef CAS.
- S. Hancock, R. Uprety, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 14819–14824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and biological protocols, supplementary figures, and copies of NMR spectra. See DOI: 10.1039/c0sc00373e |
| This journal is © The Royal Society of Chemistry 2011 |