A simple method for the synthesis of hyaluronic acid coated magnetic nanoparticles for highly efficient cell labelling and in vivo imaging†
Mohammad H.
El-Dakdouki
a,
Kheireddine
El-Boubbou
a,
David C.
Zhu
bc and
Xuefei
Huang
*ac
aDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: xuefei@chemistry.msu.edu; Fax: +1-517-353-1793; Tel: +1-517-355-9715, Ext. 329
bDepartments of Radiology and Psychology, Michigan State University, East Lansing, Michigan, 48824, USA
cBiomedical Imaging Research Centre, Michigan State University, East Lansing, Michigan, 48824, USA
First published on 7th October 2011
Abstract
Highly stable colloidal hyaluronic acid coated magnetic nanoparticles were prepared via a ligand exchange method. These particles exhibited excellent cell labeling efficiencies and superior potential as MRI contrast agents, which are useful to target tumor cells expressing hyaluronic acid receptors such as CD44.
Magnetic nanoparticles (MNPs) are undergoing intensive studies for various biomedical applications.1–4 A prerequisite for successful in vivo usage of MNPs is their stability, especially in the presence of serum proteins. Furthermore, it is desirable that the nanoparticles (NPs) can be guided to desired cells. To accomplish this, the surface of the MNPs can be modified with targeting ligands,5 an example of which is hyaluronic acid (HA). HA is a naturally occurring poly-anionic polysaccharide, which plays important roles in wound healing, cell differentiation, proliferation, and migration.6 In cancer, the interaction between HA and its receptors such as CD44, regulates tumor growth, metastasis and multi-drug resistance.7,8 There are high current interests in developing new synthetic methods for HA-coated MNPs and exploring their biological applications.9–13 In order to immobilize HA, the NPs can be functionalized first with amines followed by amide coupling with the carboxylic acid moieties in HA.9,10 Alternatively, HA can be derivatized with dopamine, dihydrazide, or a hydrophobic moiety such as pyrene which then encapsulate the MNP core producing HA–NPs.11–13 All these procedures require multi-step prior functionalization of either the NP or HA, which are time consuming. Derivatization of HA may also adversely affect the ability of HA to bind to its receptors,14 as it is known that an intact HA decasaccharide sequence or longer is required to have high affinities with CD44.15 Whereas using native HA was desirable to retain high receptor affinity, attempts to directly attach unmodified HA on MNPs have not been successful.9,13 Herein, we report a new protocol to fabricate HA–MNPs without the need of prior HA or NP derivatization and the preliminary investigations of their biological applications.
Our synthesis started from the preparation of two types of MNP cores, i.e., cobalt ferrite (CFNP) and iron oxide magnetite (IONP) (Fig. 1a), through the thermal decomposition method.16 Both MNPs contain a hydrophobic exterior coating of oleic acid (OA) with core diameters around 6 nm. As a result, the NPs could be well dispersed in organic solvents such as chloroform, hexanes, THF, and toluene, but were insoluble in water. As HA contains multiple carboxylic groups, we hypothesized that HA should be able to directly displace OA off the surface of the MNPs, rendering them water soluble. However, the challenge is that the highly hydrophilic HA does not dissolve significantly in any organic solvent.9,13 Previously, to solubilise HA, the carboxylic acids of HA were converted to the more organic compatible quaternary ammonium salts through ion exchange reactions.17,18 Alternatively, polyethylene glycol (PEG) was added to HA to form a nano-complex, which then became more soluble in organic solvents for further derivatization.19 To simplify the process without the need for any exogenous agents, we explored a ligand exchange reaction by creating a two phase system where the IONP dispersion in THF was mixed with a basic aqueous KOH solution of HA (IONP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HA w/w 1:1). Vigorous stirring of this mixture at room temperature did not trigger the ligand exchange with no NPs detected in the aqueous layer after 48 h. Upon heating the reaction to 80 °C, a significant amount of NPs precipitated, which were no longer soluble in THF or hexanes. This was attributed to the incomplete exchange of OA to HA resulting in NP agglomeration, which was supported by dynamic light scattering measurements revealing that the particles were poly-dispersed with hydrodynamic diameters over 300 nm. In order to better coat the MNPs, the amounts of HA were increased to up to 1:3 w/w NP:HA. In other experiments, hexanes were used in the two phase system to more efficiently retain OA in the organic layer. However, none of these trials resulted in much improvement.
:HA w/w 1:1). Vigorous stirring of this mixture at room temperature did not trigger the ligand exchange with no NPs detected in the aqueous layer after 48 h. Upon heating the reaction to 80 °C, a significant amount of NPs precipitated, which were no longer soluble in THF or hexanes. This was attributed to the incomplete exchange of OA to HA resulting in NP agglomeration, which was supported by dynamic light scattering measurements revealing that the particles were poly-dispersed with hydrodynamic diameters over 300 nm. In order to better coat the MNPs, the amounts of HA were increased to up to 1:3 w/w NP:HA. In other experiments, hexanes were used in the two phase system to more efficiently retain OA in the organic layer. However, none of these trials resulted in much improvement.
 | ||
| Fig. 1 a) Synthesis of HA–IONPs. b) Pictures of OA–IONPs and HA–IONPs in a toluene–water two phase system. c) TEM of HA–IONPs (the scale bar is 10 nm). d) Hydrodynamic diameter of HA–IONPs in PBS and 10% FBS containing PBS. | ||
We reasoned that higher temperatures may facilitate the ligand exchange process. This was tested by dissolving HA in water and IONPs in toluene (HA/IONPs w/w 1:1; water/toluene v/v 2:1), followed by refluxing for 24 h. The MNPs generated by this procedure easily dissolved in water (Fig. 1b) with a hydrodynamic diameter of 54 nm, zeta potential of −50 mV, and a narrow polydispersity index (PDI) of 0.17. The HA–IONPs were thoroughly characterized proving the successful coating of HA (Fig. S2†). Thermal gravimetric analysis (TGA) showed that HA accounted for 43% weight of the NPs. The HA–IONPs exhibited high relaxivity (r2* = 340 m M−1 sec−1), which is slightly higher than that of the FDA approved MRI contrast agent Feridex (r2* = 309 m M−1 sec−1)20 (Fig. S2†). The HA–IONPs showed superior colloidal stability with no significant changes in either the size or PDI in phosphate buffered saline (PBS) and 10% fetal bovine serum (FBS) containing PBS over one month (Fig. 1d). In contrast, incubation of Feridex with 10% FBS containing culture media led to a 33% increase of PDI (Fig. S3†). The colloidal stability of HA–IONPs is presumably because the dense anionic HA coating effectively passivates the NP surface minimizing non-specific binding by serum proteins. The HA–CFNPs were prepared in a similar manner as HA–IONPs by the ligand exchange process. Besides HA, other anionic ligands such as poly(acrylic acid) and polyethylene glycol (PEG)-phosphate21 have been immobilized on IONPs yielding monodispersed, colloidal stable NPs (Fig. S2–S4†). These results highlight that our protocol can be of general use for attaching negatively charged ligands onto MNPs.
Having validated the monodispersity and colloidal stability of HA–IONPs, we studied NP uptake and targeting of CD44 expressing human ovarian cancer cell SKOV-3. The cancer cells were incubated with NPs followed by washing to remove all unbound NPs and staining with Prussian blue to visualize intracellular iron. Dose dependent intracellular accumulation of HA–IONPs was obtained with the lowest observable concentration of 1.3 μg of Fe per mL (Fig. 2a–e, 2i). Besides HA–IONPs, cellular uptakes of Feridex and HA–DESPION by SKOV-3 cells were evaluated for comparison. No iron was detected in the cells treated with Feridex even at the highest concentration tested (21 μg of Fe per ml) (Fig. 2g). HA–DESPION was another type of HA coated MNPs, which has been shown to target cells in a CD44 dependent manner.10 HA–DESPION was fabricated by a multi-step process, i.e., the iron oxide NP core was coated first with the polysaccharide dextran followed by amine functionalization and HA immobilization. Interestingly, HA–IONPs showed a much higher cellular uptake by CD44 expressing SKOV-3 cells as compared with HA–DESPION (Fig. 2f). Quantification of the Prussian blue stain demonstrated over two orders of magnitude increase of NP uptake by SKOV-3 cells using HA–IONPs (Fig. 2i). Although the exact reason is not clear, this may be because the HA coating of HA–DESPION is mixed with dextran polymer and significant amounts of HA carboxylic acids are coupled to the amines on NP, thus lowering the avidity to HA receptors. These results highlight the importance of NP structures for mediating cellular uptake.
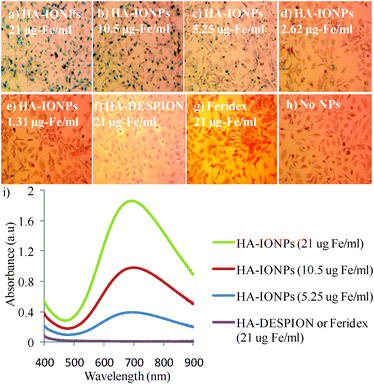 | ||
| Fig. 2 Prussian blue staining of SKOV-3 cells upon incubation with a) 21; b) 10.5; c) 5.25; d) 2.62; e) 1.31 μg of Fe per mL of HA–IONPs; f) 21 μg of Fe per mL of HA–DESPION; and g) 21 μg of Fe per mL of Feridex after washing off the unbound NPs. h) Cells without NP incubation. i) UV-vis spectra of SKOV-3 cells after incubating with various concentrations of NPs followed by washing off the unbound NPs and Prussian blue staining. | ||
The high cellular uptake coupled with high magnetic relaxivity of HA–IONPs indicated their potential utility in cell labelling and tracking by MRI. To assess this possibility, after HA–IONP incubation and removal of unbound NPs, the SKOV-3 cells were resuspended in agarose gel and T2 and T2*-weighted MR images were collected. Even with just 2.6 μg of Fe per mL of NPs, clear changes were observed in both the T2 and T2* weighted images (Fig. 3a,b and Fig. S5†), suggesting HA–IONP loaded cells can be easily tracked by MRI. In comparison, Feridex did not generate any MRI signal changes up to 21 μg of Fe per mL (Fig. 3a,b and Fig. S5†), which was consistent with the Prussian blue assay results. The HA–IONPs did not influence the viability of the cells. Following 24 h incubation with HA–IONPs, a MTS cell viability assay demonstrated that the NPs were not toxic to SKOV-3 with iron concentrations up to 49.6 μg ml−1 (Fig. 3c).
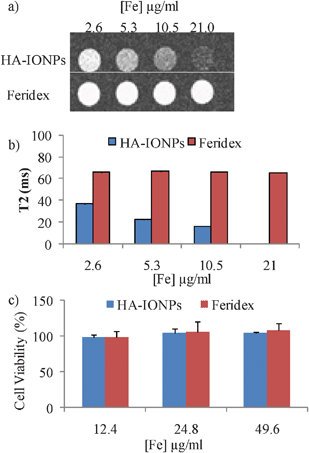 | ||
| Fig. 3 a) T2 weighted images of SKOV-3 cells upon incubation with various concentrations of HA–IONPs and Feridex after removing unbound NPs. b) T2 values of SKOV-3 cells upon incubation with various concentrations of HA–IONPs and Feridex after removing unbound NPs. The T2 value at an Fe concentration of 21 μg mL−1 was too small (<10 ms) to be quantified accurately. All experiments were repeated at least three times. The error bars were too small to be seen in these graphs. c) MTS cell viability assays showed that HA–IONPs are highly biocompatible. | ||
To investigate the utility of HA–IONPs as drug carriers, we immobilized fluorescein isothiocyanate (FITC) as a model drug (FITC–HA–IONPs). FITC was linked with adipic dihydrazide (ADH),22 which was then conjugated to HA. The toluene/water two phase ligand exchange protocol was employed to coat FITC–HA on the surface of the IONPs, producing FITC–HA–IONPs 68 nm in size with a narrow PDI (0.13) and high T2 relaxivity (r2* = 314 m M−1 sec−1). The UV-vis and fluorescence properties of FITC on NPs remained intact indicating that the FITC cargo was not affected by the experimental conditions. Fluorescence activated cell sorting studies showed significant cellular uptake of FITC–HA–IONPs by CD44 expressing cancer cells following NP incubation. The cellular uptake was significantly reduced when the FITC–HA–IONP incubation was carried out in the presence of free HA affirming the importance of HA in mediating cellular uptake (Fig. S6†).
We evaluated next the potential in vivo applications of HA–IONPs by administering HA–IONPs (0.23 mg Fe) to live mice via retro-orbital injection followed by MRI. No adverse effects upon NP administration were observed. The T2 weighted MR images of the mouse exhibited significant signal losses in several organs including the kidneys and liver (Fig. 4). The darkening in T2 weighted images suggested extensive accumulation of HA–IONPs in these organs. This could be due to the presence of HA receptors such as lymphatic endothelium-specific hyaluronan receptor (LYVE-1) in the kidneys and HA receptor for endocytosis (HARE) in the liver,23,24 as well as uptake by the reticuloendothelial system.
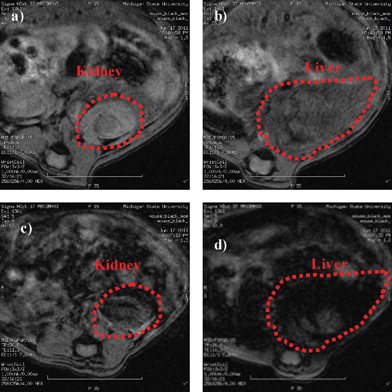 | ||
| Fig. 4 T2 weighted images of a) kidney and b) liver of a live mouse prior to injection of HA–IONPs; c) kidney and d) liver of the mouse after injection of HA–IONPs. | ||
In conclusion, we describe a simple method for the preparation of HA coated IONPs without prior functionalization of HA or NPs, which can be applied to the immobilization of other water soluble anionic ligands as well as magnetic nanoparticles such as cobalt ferrite. The colloidal stable and biocompatible HA–IONPs prepared using this protocol are much more efficient in labelling CD44 containing tumor cells compared to other types of MNPs. HA–IONPs present a useful platform for targeted drug delivery, cellular tracking and in vivo imaging of HA receptor expressing organs and tissues such as tumor, kidneys, liver, and inflammatory sites.
Acknowledgements
This work was partially supported by an NSF CAREER award and the National Cancer Institute (R01-CA149451) (XH). We would like to thank the Department of Radiology, Michigan State University for the very generous support towards access of the MRI scanner.References
- K. El-Boubbou, D. C. Zhu, C. Vasileiou, B. Borhan, D. Prosperi, W. Li and X. Huang, J. Am. Chem. Soc., 2010, 132, 4490–4499 CrossRef CAS and references cited therein..
- Y.-W. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- C. Corot, P. Robert, J.-M. Idee and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS.
- A. K. Gupta, R. R. Naregalkar, V. D. Vaidya and M. Gupta, Nanomedicine, 2007, 2, 23–39 CrossRef CAS.
- L. Lapcik, Jr., L. Lapcik, S. De Smedt, J. Demeester and P. Chabrecek, Chem. Rev., 1998, 98, 2663–2683 CrossRef.
- S. Misra, P. Heldin, V. C. Hascall, N. K. Karamanos, S. S. Skandalis, R. R. Markwald and S. Ghatak, FEBS J., 2011, 278, 1429–1443 CrossRef CAS.
- A. Bartolazzi, R. Peach, A. Aruffo and I. Stamenkovic, J. Exp. Med., 1994, 180, 53–66 CrossRef CAS.
- H.-J. Chung, H.-S. Lee, K. H. Bae, Y.-H. Lee, J.-N. Park, S.-W. Cho, J.-Y. Hwang, H.-W. Park, R. Langer, D. Anderson and T.-G. Park, ACS Nano, 2011, 5, 4329–4336 CrossRef CAS.
- M. Kamat, K. El-Boubbou, D. C. Zhu, T. Lansdell, X. Lu, W. Li and X. Huang, Bioconjugate Chem., 2010, 21, 2128–2135 CrossRef CAS.
- Y. Lee, H. Lee, Y. B. Kim, J. Kim, T. Hyeon, H. Park, P. B. Messersmith and T. G. Park, Adv. Mater., 2008, 20, 4154–4157 CAS.
- A. Kumar, B. Sahoo, A. Montpetit, S. Behera, R. F. Lockey and S. S. Mohapatra, Nanomed.: Nanotechnol., Biol. Med., 2007, 3, 132–137 CrossRef CAS.
- E.-K. Lim, H.-O. Kim, E. Jang, J. Park, K. Lee, J.-S. Suh, Y.-M. Huh and S. Haam, Biomaterials, 2011, 32, 7941–7950 CrossRef CAS.
- J. Kim, K. S. Kim, G. Jiang, H. Kang, S. Kim, B.-S. Kim, M. H. Park and S. K. Hahn, Biopolymers, 2008, 89, 1144–1153 CrossRef CAS.
- R. Tammi, D. MacCallum, V. C. Hascall, J. P. Pienimäki, M. Hyttinen and M. Tammi, J. Biol. Chem., 1998, 273, 28878–28888 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- M. Zhang and S. P. James, Polymer, 2005, 46, 3639–3648 CrossRef CAS.
- Y. Luo and G. D. Prestwich, Bioconjugate Chem., 1999, 10, 755–763 CrossRef CAS.
- H. Lee, K. Lee and T. G. Park, Bioconjugate Chem., 2008, 19, 1319–1325 CrossRef CAS.
- Y.-X. J. Wang, S. M. Hussain and G. P. Krestin, Eur. Radiol., 2001, 11, 2319–2331 CrossRef CAS.
- U. I. Tromsdorf, O. T. Bruns, S. C. Salmen, U. Beisiegel and H. Weller, Nano Lett., 2009, 9, 4434–4440 CrossRef CAS.
- B. Ahn, S. G. Rhee and E. R. Stadtman, Anal. Biochem., 1987, 161, 245–257 CAS.
- H. W. Lee, Y. X. Qin, Y. M. Kim, E. Y. Park, J. S. Hwang, G. H. Huo, C. W. Yang, W. Y. Kim and J. Kim, Cell Tissue Res., 2011, 343, 429–444 CrossRef CAS.
- J. A. Weigel, R. C. Raymond, C. McGary, A. Singh and P. H. Weigel, J. Biol. Chem., 2003, 278, 9808–9812 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures and images. See DOI: 10.1039/c1ra00737h |
| This journal is © The Royal Society of Chemistry 2011 |