Copper(I) speciation in mixed thiosulfate-chloride and ammonia-chloride solutions: XAS and UV-Visible spectroscopic studies†
Barbara E.
Etschmann
ab,
Jay R.
Black
cd,
Pascal V.
Grundler
ab,
Stacey
Borg
e,
Dale
Brewe
f,
D. C.
McPhail
g,
Leone
Spiccia
*c and
Joël
Brugger
*ab
aSchool of Earth and Environmental Sciences, The University of Adelaide, 5000 Adelaide, Australia
bDivision of Mineralogy, South Australian Museum, North Terrace, 5000 Adelaide, Australia
cSchool of Chemistry and Centre for Green Chemistry, Monash University, 3800 Vic, Australia
dInstitute for Geophysics and Planetary Physics, University of California, Los Angeles, 90095, CA, USA
eCSIRO Exploration & Mining, Clayton, Victoria, Australia
fAdvanced Photon Source, Argonne National Laboratory, 9700 S. Cass Ave, Argonne, IL 60439, USA
gResearch School of Earth Sciences, The Australian National University, ACT 0200, Australia. E-mail: Joël Brugger: joel.brugger@adelaide.edu.au, Leone Spiccia: leone.spiccia@monash.edu; Fax: +61 8 8303 6222, +61 3 9905 4597; Tel: +61 8 8207 7451, +61 3 9905 4526
First published on 19th October 2011
Abstract
Thiosulfate and ammonia mixtures may be more environmentally benign alternatives to cyanide for leaching Au from ores. In this method, the Cu(I)/Cu(II) couple acts as a redox mediator aiding in the oxidative dissolution of metallic Au. Information about the speciation of Cu(I) and Cu(II) in these lixiviant solutions is paramount to the optimization of gold ore processing conditions. With this in mind, we have carried out XANES, EXAFS and UV-Vis spectroscopic studies of the speciation of Cu(I) in mixed thiosulfate-chloride and ammonia-chloride solutions. In thiosulfate-chloride solutions, the EXAFS studies indicate that the geometry of the predominant Cu(I) complex is distorted trigonal (triangular planar), with an average of 2 sulfur atoms + 1 oxygen atom occupying the coordination sphere. This indicates that the stability of the [Cu(S2O3)3]5− complex is lower than previously proposed. Formation constants for Cu(I) thiosulfate complexes have been derived on the basis of systematic UV-Vis measurements of solutions with varying [S2O3]/[Cl] ratios. Only one mixed chloride-thiosulfate complex, [Cu(H2O)(S2O3)Cl]2−, was found to predominate over the range of conditions investigated. For Cu(I) in ammonia-chloride solutions, our results confirm the broad stability of [Cu(NH3)2]+ and we have also identified a stable mixed amminechlorocopper(I) complex, [CuCl(NH3)]+. XAS reveals that these two complexes share a linear geometry. This study demonstrates that combinations of methods are required to decipher the geometry and thermodynamic properties of transition metal complexes in mixed ligand chemical systems where many species may coexist. Our results allow more comprehensive predictions of solution speciation and contribute to efforts to design improved methods to process gold ore with thiosulfate and ammonia lixiviants.
1. Introduction
Gold is typically extracted using the cyanidation/carbon-in-pulp process, a method that is effective and economical when applied to ores containing relatively low gold concentrations (0.5–3 ppm Au).1 Increasing pressure from environmental protection agencies and public perception, which frown upon cyanide due to its toxicity and potential volatility, has resulted in the investigation of more benign lixiviants, such as thiosulfate and ammonia.2,3 In these alternative methods, the copper(I)/(II) couple acts as a redox mediator aiding in the oxidative dissolution of gold. Metallic Au is oxidized by Cu(II), and the resulting Cu(I) can be oxidized back to Cu(II) by reaction with atmospheric oxygen to complete the catalytic cycle. In addition, since Cu(II) can also oxidize thiosulfate to tetrathionate, an understanding of the solution speciation and stability of individual species is important for decreasing the amount of reactant needed during processing.2 The chemistry of the ammonia–thiosulfate–copper(I)/(II) system is complicated due to the presence of two complexing ligands, ammonia and thiosulfate. Given that gold processing plants in arid and semi-arid regions (e.g., Australia) may need to use highly saline local groundwaters, it is also important to investigate the effect of chloride (e.g., “Water Management at KCGM”4). Copper(I) speciation is important in the gold oxidation cycle and, thus, there is a need to understand the nature and properties of Cu(I) complexes that form in these chemical environments, in order to design new hydrometallurgical methods that optimize leaching parameters, extract gold more efficiently and quickly, and minimize the amounts of lixiviants.The complexation of Cu(I) by chloride ligands from ambient temperature to 500 °C has been the subject of recent solubility and spectroscopic studies.5–11 The consensus emerging from these studies is that the linear complexes [H2O–Cu–Cl]0 and [Cl–Cu–Cl]− dominate the speciation of Cu(I) in acidic chloride brines. The trigonal planar [CuCl3]2− ion exists at low temperature (< 200 °C) and high [Cl−] (> 2 m), but detailed XAS studies showed that the tetrahedral complex [CuCl4]3−, commonly found in solids, does not exist in solution,5 a result independently corroborated by ab initio molecular dynamic simulations.12 Herein, the thermodynamic constants for Cu(I) chloro-complexes from Brugger et al.5 are used.
Fewer data are available for thiosulfatocopper(I) complexes, and little information is available on their geometry. According to Smith and Martell,13 [Cu(S2O3)]− predominates over a broad range of S2O32− activities (e.g., up to ∼16 mM S2O32− in a 1 mM Cu+ solution at pH 9.25), whereas [Cu(S2O3)2]3− and [Cu(S2O3)3]5− form at higher [S2O32−] (Fig. 1). Only two studies report formation constants for [Cu(S2O3)]−,14,15 and they are in relatively poor agreement (log β = 8.91/9.91 and 10.35, respectively; Table 1). The study of Golub et al.14 does not provide sufficient details on the reaction conditions (e.g., metal ion concentrations are not reported), and hence this study does not meet the quality criteria outlined by Martell and Motekaitis,16 so we cannot assess the reliability of their results. A good consensus exists for the formation constant of [Cu(S2O3)2]3−, with three of four studies in broad agreement (11.69 < log β < 12.30;15,17,18; Table 1). Two studies15,19 provide a consistent formation constant for [Cu(S2O3)3]5− (13.64 < log β < 14.30), which is much higher than the log β = 10.35/11.75 reported by Golub et al.14 (Table 1). Only one study has investigated mixed chloride-thiosulfate complexes. Using potentiometric methods, Boos and Popel20 proposed that [Cu(S2O3)2Cl]4− was the predominant species present for [Cl−] from 0.01 to 0.1 M and [S2O32−] from 1.5 to 5.4 mM, which are reflected in Fig. 1a.
 | ||
| Fig. 1 Distributions of Cu(I) species in the thiosulfate-ammine-chloride system. Full circles and number labels refer to solution compositions from Tables S1 and S3. Activity of Cu species 10−3, T = 25 °C. | ||
| Complex | Stability constants (Log β) | Ionic strength (m) | Medium | References |
|---|---|---|---|---|
| a N.B. Golub et al.43 also reported formation constants of 11.5/12.38 for [Cu(S2O3)47− in Na2SO4/K2SO4. b Preferred value in Smith and Martell.13 c Ka for the reaction [Cu(NH3)2]+ + NH3(aq) = [Cu(NH3)3]+. d Solis et al.21 also report minor amounts (< 15%) of other mixed ligand complexes, and hence impossible to characterize precisely. e Cited by Black.49 | ||||
| Cu(I) chloride complexes | ||||
| CuCl0 | 4.17 | 0 | NaCl | 5 |
| CuCl2− | 5.46 | 0 | NaCl | 5 |
| CuCl32− | 4.75 | 0 | NaCl | 5 |
| Cu(I) thiosulfate complexes | ||||
| Cu(S2O3)− | 10.35b | 0.1–7 | Na2SO4 | 15 |
| 8.91/9.91 | Na2SO4/K2SO4 | 43 | ||
| 8.5 | 0 | Recommended.This study | ||
| Cu(S2O3)23− | 12.27b | 1.6 | Na2SO4 | 15 |
| 12.30 | n.r. | n.r. | 18 | |
| 11.69 (18 °C) | Variable | Variable | 17 | |
| 9.32/10.6 | Na2SO4/K2SO4 | 43 | ||
| 12.0(5) | 0 | Recommended. This study | ||
| Cu(S2O3)35−a | 14.30 | 1.2 | KNO3 | 19 |
| 13.70b | 1.6 | Na2SO4 | 15 | |
| 13.64 | 1.0 | Na2SO4 | 15 | |
| 10.34/11.75 | Na2SO4/K2SO4 | 43 | ||
| 9.9(5) | 0 | Recommended. This study | ||
| Cu(I) mixed thiosulfate-chloride complexes | ||||
| Cu(S2O3)2Cl4− | 12.89 | n.r. | n.r. | 20 |
| Cu(S2O3)Cl23− | 7.63(15) | 0 | Recommended. This study | |
| Cu(S2O3)Cl2− | 9.61(15) | 0 | Recommended. This study | |
| Cu(I) ammine complexes | ||||
| Cu(NH3)+ | 5.74b | 2.0 | NH4NO3 | 44 |
| 5.80 | 3.0 | (NH4)2SO4 | 45 | |
| Cu(NH3)2+ | 9.92b | 0.1 | KNO3 | 46 |
| 11.38 | 1 | NaClO4 | 21 | |
| 10.7(+ 0.2/−0.5) | 0 | Recommended. This study | ||
| Cu(NH3)3+ | Ka = ∼0.04 | 0.5 | NaClO4 | 47 |
| Ka = 0.05 | 1.0 | NaClO4 | 48 | |
| Ka = 0.05 | 3.0 | (NH4)2SO4 | 45 | |
| ∼10.5 | 0 | Recommended. This study | ||
| Cu(I) mixed ammine-chloride complexes | ||||
| Cu(NH3)Cl0 | 8.92 | 1.0 | NaClO4 | 21 |
| 8.38(+ 0.2/−0.3) | 0 | Recommended. This study | ||
Investigations of speciation in the Cu(I)–NH3 system (Table 1) indicate that [Cu(NH3)2]+ is the main amminecopper(I) complex, and that [Cu(NH3)]+ and [Cu(NH3)3]+ play only minor roles (Fig. 1). A single study (Solis et al.21) examined the presence of mixed chloro-ammine complexes by polarography and concluded that the most abundant complex is [Cu(NH3)Cl]0, although the data suggested that other minor mixed complexes are present (e.g., [Cu(NH3)Cl2]−).
The formation constants for Cu(I)–S2O32− and Cu(I)–NH3 species are approximately 4–8 orders of magnitude larger than those for the Cu(I)–Cl− complexes (Table 1); hence even in chloride-rich solutions (e.g. 1 m Cl−; Fig. 1), Cu(I) speciation is expected to be dominated by ammine and thiosulfato complexes. This study aims to investigate the significance of mixed chloride complexes in the Cu(I)–Cl−–S2O32− and Cu(I)–Cl−–NH3 systems, to characterize the composition and geometry of the predominant ammine and thiosulfato complexes using a combination of X-ray Absorption Spectroscopy (XAS) and UV-Vis spectroscopy, and to provide a reliable set of formation constants for Cu(I) complexes that is consistent with existing information for Cu(I) complexation in the Cl−–NH3–S2O32− system. This will provide more comprehensive numerical models for these complex chemical systems, which can be used in the refinement and optimization of methods for extracting gold.
2. Experimental methods and data analysis
2.1 Starting materials and solutions preparation
Freshly synthesized copper(I) chloride was dissolved with the requisite amounts of sodium chloride, sodium thiosulfate, ammonium chloride and sodium hydroxide in water to achieve the solution compositions listed in the supplementary data Tables S1–S4.† All chemicals were of analytical-grade or better and were used as received. All solutions were prepared by weighing and, to minimize oxidation of Cu(I) to Cu(II), they were prepared from double-deionized water degassed of oxygen by boiling and bubbling nitrogen gas through the solution for at least 15 min, immediately before preparation in a glove box under nitrogen atmosphere. Samples were then loaded, in the glove box, into either stopped PTFE solution cells that were used to obtain XAS data under ambient conditions or stoppered quartz cuvettes for UV-Vis spectrophotometry. Stock solutions of copper(I) chloride were filtered through Millipore 0.22 μm membrane filters before mixing with ammonia and thiosulfate stocks. Identical solutions could not be measured for both the XAS and UV-Vis experiments, due to the higher Cu(I) concentration and consequently higher S2O32− and NH3 concentrations (in order to prevent precipitation of copper compounds) required for the XAS experiments, which resulted in covering a different range of compositions to that for the UV-Vis experiments. The pH values of stocks and samples containing ammonia were measured outside of the glove box using a glass electrode calibrated with freshly prepared sodium tetraborate (pH25 °C 9.2) and phosphate (pH25 °C 7.2) buffer solutions.22 The pH of thiosulfate solutions was not adjusted and was self-buffered at ∼5.5. Copper(I) hydrolysis is not expected to be important as the pH of the solutions was well below the first hydrolysis constant for Cu(I) (log K [Cu(OH)]0(aq) = 11.36 at 25 °C).10,23 Thus, under these experimental conditions, the speciation and thermodynamic properties could be determined whilst minimising the impact of mixed hydroxide complexes.2.2 UV-Vis measurements and interpretation
Spectrophotometric measurements were carried out at 25 °C using a Varian CARY® 5G UV-Vis-NIR instrument equipped with a water-regulated temperature controller. The sample solutions were placed in 1 cm path length quartz cuvettes in the nitrogen glove box, and sealed with a screw on cap and Teflon o-ring. For each solution, the baseline spectrum of a solution with the same composition but containing no Cu was collected before measuring the spectrum of the Cu-bearing solution. The baseline spectrum was subtracted from the spectrum of each sample solution to correct for detector response, and for the absorption of the cell and solution matrices. The absorbance of each solution was measured at 1 nm increments between 1200 and 200 nm. It was typically in the range of 0–2.5 absorbance units, with a reproducibility of 0.01 units.Solution UV-Vis-NIR spectrophotometry measures a variety of electronic transitions including those between orbitals on the metals and ligands (and vice versa) and the complexes and solvent. It can therefore provide information about the electronic structure of the complex, e.g., the oxidation state of the metal, the geometry of the complex, and the nature of the ligands. The intensity of electronic transitions (molar absorbances ∼1000–5000 M−1 cm−1) occurring in the 210–320 nm region for the Cu(I) system indicates that these bands originate from charge transfer transitions. Systematic measurements of UV-Vis spectra for solutions containing varying amounts of ligands were used to identify the complexes present in the experiments and derive their apparent formation constants, providing a test of the validity of proposed speciation models. The interpretation is based on the Beer–Lambert law, which relates the absorbance (A) at a given wavelength (λ) to the molar absorptivity (εi,λ) and the molar concentrations (Mi) of each of the i absorbing species present in solution:
 | (1) |
When considering more than one wavelength, the Beer–Lambert law (eqn 1) can be written in a matrix form as:
| A = C*E | (2) |
On the basis of preliminary analysis and taking into account available literature information, a chemical speciation model can be established. The final quantitative analysis is conducted using non-negativity constraints for the molar absorptivity coefficients, and a complete speciation model to calculate the matrix C (i.e., the elements of C are constrained by mass balance and mass action equations). This analysis delivers a set of equilibrium constants (Log K) for the formation of the different Cu(I) complexes. For the distribution of species calculations, activity coefficients were estimated with the “b-dot” equation,26 using the b-dot parameters listed for NaCl solutions from Helgeson and Kirkham.27 The ion size parameters were set to 5 Å for Cu(I)-complexes, 9 Å for H+, and 4 Å for all other charged ions and complexes.28 The ionic strength of solutions ranged from ∼0.5 to 5 molal (Tables S3 and S4†), which is within the limit for the b-dot model description of NaCl solutions.24,29
2.3 X-ray absorption spectroscopy measurement and analysis
XANES spectra for a given stoichiometry and geometry of Cu(I) thiosulfato and ammine complexes were calculated ab initio using the FDMNES package developed by Joly,34 following the procedure outlined in Brugger et al.5 and Testemale et al.35. The final states and resulting absorption cross sections were calculated using the Finite Difference Method (FDM) to solve the Schrödinger equation in the cluster. This method allows a totally free potential shape, thus avoiding the limitations imposed by the Muffin Tin approximation. Compared to methods that rely on the Muffin Tin approximation, FDM is of particular interest in the case of low symmetry and/or non-dense structures, as in the case of the low-symmetry aqueous complexes considered here.5,34
Brugger et al.5 calculated the XANES spectra for linear, trigonal planar and tetrahedral Cu(I)-chloro complexes. A comparison between calculations for the crystalline solids cubic Cu2O(s) and cubic CuCl(s) performed with different cluster sizes demonstrated that there are only minor differences between calculations that include only the first coordination shell and those that include atoms beyond the first shell in the case of ‘dense’ coordination, as is the case for CuCl(s) in which the Cu is tetrahedral and coordinated by four chloride ions. However, in the case of more open structures, such as Cu2O(s) in which Cu is linear and coordinated by two oxygen atoms, inclusion of second-shell atoms resulted in significant improvements between the calculated and experimental spectra of the crystalline compound.
The raw calculations represent the photo-absorption cross-section related to the transition amplitudes between initial and final states. To make these calculated spectra comparable with the experimental spectra, these calculations are convoluted with a Lorentzian function that has an energy-dependent width in order to reproduce the core-hole lifetime broadening (1.55 eV for the Cu K-edge36) and the inelastic plasmon interactions with the photoelectron, and with a Gaussian function to reproduce the experimental resolution, 1.26 eV in this case (Cu K-edge × ΔE/E = 8979 × 1.4 × 10−4). Note that these values are set a priori and not fitted in the subsequent analyses, and that the calculated peak shapes tend to be sharper than the experimental data as thermal effects and the inelastic mean free path are not taken into account.37
The atoms were kept neutral for all the calculations and the evolution of the charges on the atoms was not studied, as the experimentally observed changes are essentially structural. Atomic charges need to be considered in the case of a stable structure that changes very little during the experiment and in which the evolution of charges is likely to be detectable.38,39 In the present study, however, the XANES calculations are used essentially to confirm the nature (i.e., geometry and stoichiometry) of the main Cu species present in the aqueous solutions.
3. Results and discussion
The determination of a speciation model that incorporates all the available data (current measurements as well as published results) is an iterative process, where the results of one experiment, or previously published study, are used in the analysis of the other datasets until a self-consistent model is obtained. In the following sections, we first present the qualitative XANES analysis and ab initio simulation of the XANES spectra to identify the possible geometries and, thus, the allowable stoichiometries of the species. This is followed by the analysis of EXAFS data for ‘end-member’ solutions, i.e., solutions containing the most contrasting speciation, usually corresponding to solutions with, for example, the least and most amounts of chloride. Finally, the systematic UV-Vis spectral measurements are used to confirm the nature of the species present and to retrieve their formation constants. The results for the thiosulfate system are presented first followed by those for the ammonia system.A note regarding notation: because the information from XAS solution data is generally limited to first shell/nearest neighbors interactions, only those atoms involved in the simulation or fit will be indicated within parentheses, i.e., (CuSClO). For fitting the UV-Vis data and for the thermodynamic interpretation, we use a stoichiometric notation for the entire complex, which will be indicated by square brackets, i.e., [Cu(S2O3)Cl(H2O)]3−. Finally, we follow the usual convention in thermodynamic modeling that excludes coordinated water molecules from the species names; no parenthesis is used for this notation, e.g. Cu(S2O3)Cl2−.
3.1 The Cu–S2O3–Cl system
XANES spectra for all solutions are broadly similar (Fig. 2), the main difference being an increase in the intensity of the pre-edge with increasing S2O32−/Cu ratio. Solutions with the same S2O32−/Cu ratio and varying chloride concentrations have very similar XANES spectra; i.e., for these compositions the effect of chloride is small. The XANES features strongly suggest that Cu(I) exists predominantly as three-coordinate complexes.33 A linear structure can be excluded on the basis of the low intensity of the pre-edge (see [Cl–Cu–Cl]−,5 [HS–Cu–SH]−42), as can a tetrahedral geometry because of the lack of a shift in the position of the pre-edge to higher energy.5,33 The differences between the spectra shown in Fig. 2 result from either structural distortions and/or variation in the ligands occupying the Cu(I) coordination sphere, but not from a change in coordination geometry.

 | ||
| Fig. 3 XANES simulations (raw spectra only) of the Cu–S2O32−–Cl− system, compared with experimental data. (a) Effect of changing geometry. (b) Effect of changing distance. (c) Effect of ligand substitution. | ||
Fig. 3a illustrates the effect of geometry on the XANES spectra. XANES simulations demonstrate that both end-member solutions can be modeled well using a distorted trigonal planar geometry. When the atoms are arranged in an equilateral triangle (symmetry C3), the peaks in the resulting calculated spectrum do not match the positions of the peaks in the experimental spectra. In contrast, a close agreement was found between observed and calculated spectra for a near-T-shaped geometry or a Y-shaped geometry (Fig. 3a). Slight distortion of the T-shaped geometry (i.e. rotating one or more atoms by 5°) results in a decrease in the intensity of the second peak; this small effect virtually disappears when the spectra are convoluted (i.e., broadened by the experimental resolution) (Fig. 3a).
All the raw calculated spectra have a small peak, indicated with dashed line 2 in Fig. 3a, between the two main large peaks (indicated with 1 and 3). This small feature was not observed in the measured spectra, but this is probably an effect of experimental resolution; indeed this feature was absent when the spectra were convoluted with an experimental broadening routine (Fig. 3a). The shift in position of peak ‘3’ can also be accounted for by slight changes in S bond lengths (Fig. 3b). For example, for the (CuS2O) entity, an increase in the Cu-S distance from 2.20 to 2.25 Å causes a 0.3 eV shift in the peak 3 position for the T- and Y-shaped geometries (Fig. 3b). A change in Cu–O bond length from 2.0 to 2.1 Å (leaving the Cu–S distance at 2.25 Å) does not affect the position of peak ‘3’, but slightly drops its intensity, as well as shifting the position of the minor peak ‘2’.
Fig. 3c shows the effect of substituting O, S and Cl ligands. Although the spectra shown are for a T-shaped geometry, the trends are the same for Y-shaped geometries. Distances used for these calculations were Cu–O = 2.0 Å, Cu–Cl = 2.25 Å and Cu–S = 2.25 Å, which were optimized from XANES FDMNES calculations and are similar to those determined by EXAFS (see section 3.1.3). The change in ligand is reflected mainly by a shift in the position of the peak number ‘3’; a similar shift is observed in the experimental data. The spectra that least resemble the experimental spectra are those containing more than one oxygen atom in the coordination sphere (i.e., (CuO2S), (CuO2Cl), (CuO3)). All the other calculated spectra have four peaks within the first 20 eV, labeled by the numbers ‘1’ to ‘4’ on Fig. 3c. Peak no. ‘3’ is at a too low energy in the calculated (CuS3), (CuCl3), and (CuCl2S) spectra (i.e., models with longer bond lengths). However, the spectra calculated for complexes with one oxygen ligand are close to the experimental data. The main change between the two experimental spectra is a shift of the peak number ‘3’ to lower energy from solution CuS2O3_09 to solution CuS2O3_06. Such a shift can be explained by a change from (CuS2O) to (CuCl2O), but is also consistent with a change from (CuS2O) to (CuSClO).
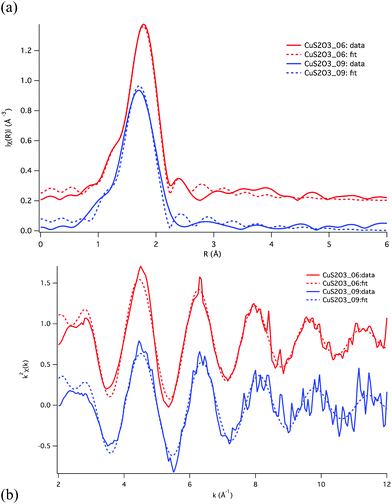 | ||
| Fig. 4 EXAFS fits for the Cu–S2O32−–Cl− system, shown in R-space (a) and k-space (b). The fit parameters are listed in Table 2. Fits were conducted simultaneously on both the CuS2O3_06 and CuS2O3_09 solutions, and are based on the assumption that speciation is dominated by trigonal complexes. | ||
| Model | donor | N | R (Å) | δ 2 (Å2) | ΔE0 (eV) | χ2red | |
|---|---|---|---|---|---|---|---|
| a Constrained to be the same for the Cl and N atoms. | |||||||
| Solution CuS2O3_ | |||||||
| 06 | Trigonal | S | 2.1(3) | 2.24(1) | 0.006(3) | 7(2) | 25.8 |
| O | 0.9(3) | 2.11(4) | 0.009(20) | ||||
| 09 | Trigonal | S | 1.8(3) | 2.20(3) | 0.006(3) | 7(2) | 25.8 |
| O | 1.2(3) | 2.10(5) | 0.009(20) | ||||
| 06 | Trigonal | Cl | 1.9(3) | 2.223(9) | 0.005(2) | 7(2) | 35.4 |
| O | 1.1(3) | 2.13(5) | 0.026(23) | ||||
| 09 | Trigonal | S | 1.6 (fix) | 2.21(2) | 0.005(2) | 7(4) | 23.9 |
| O | 1.4 (fix) | 2.09(4) | 0.019(22) | ||||
| Solution CuNH3_ | |||||||
| 01 | linear | N | 2 | 1.90(1) | 0.002(1) | 2(1) | 39 |
| 09 | Trigonal | Cl | 2.1(4) | 2.27(32) | 0.04(15) | 5(2) | 12.7 |
| N | 0.9(4) | 2.21(1) | 0.008(3) | ||||
| 09 | Trigonal: 50% | Cl | 3 (fix) × 0.50 | 2.219(9) | 0.004(1) | 0(1) | 32 |
| linear: 21% | Cl | 1 (fix) × 0.21 | 2.10(2) | 0.003(2)a | |||
| N | 1 (fix) × 0.21 | 1.94(10) | 0.003(2)a | ||||
| linear: 21% | Cl | 2 (fix) × 0.21 | 2.10(2) | 0.003(2)a | |||
| linear: 7% | N | 2 (fix) × 0.07 | 2.14 (fix) | 0.003(2)a | |||
These XAS results suggest some discrepancy with the speciation predicted using available thermodynamic properties (Fig. 1a; Table 1). The XANES data unambiguously reveal that the copper(I) geometry and ligation change little over the range of conditions investigated. This result is confirmed by EXAFS that reveals that the average coordination in solutions CuS2O3_09 and _06 consists of ∼2 (Cl + S) and ∼1 oxygen, in contrast to the transition from 1S + 2O (i.e. [Cu(S2O3)(H2O)2]−) (solution 9) to 2S + 1Cl (i.e. [Cu(S2O3)2Cl]4−) (solution 06) inferred using the available thermodynamic properties. In the following section a speciation model for Cu(I) in mixed thiosulfate-chloride solutions will be developed that is consistent with the XAS data and the systematic UV-Vis data.
The bond lengths determined from EXAFS compare well with bond lengths published in the literature. As can be seen in Table 3, Cu(I) coordinated to two ligands, either sulfur or chlorine have similar bond lengths (∼2.15 Å). Similarly Cu(I) coordinated to three sulfur or chlorine ligands has a longer bond length around 2.27 Å, and Cu(I) in a four-fold coordination with sulfur or chlorine has a bond length around 2.35 Å. This indicates that the Cu-S bond length is influenced by the number of coordinating atoms. In the context, the Au(I)-S distance measured by EXAFS on Au(I)-hydrosulfide solutions under hydrothermal conditions is similar to the Au(I)-S distance found in solids with similar (linear) coordination geometry (Table 3). As expected, the Cu(I)–O and Cu(I)–N bond lengths are shorter than Cu(I)–S or Cu(I)–Cl bond lengths (Table 2). In summary, the bond lengths determined from EXAFS refinements are consistent with a three-fold coordination around the Cu(I) atom.
| Structure | Coordination number | Shortest Bond length | Ref. |
|---|---|---|---|
| a Solid state structure. b Solution. | |||
| Copper(I) | |||
| Na4(Cu(NH3)4)–(Cu(S2O3)2)2 | Four, Cu(I)–S (S from thiosulfate) | 2.357 Åa | 50 |
| CuFeS2 (chalcopyrite) | Four, Cu(I)–S | 2.279 Åa | 51 |
| CuCl (nantokite) | Four, Cu(I)–Cl | 2.345 Åa | 52 |
| K5Cu(S2O3)3.(2.25)H2O | Three, Cu(I)–S | 2.225(1) Åa | 62 |
| 2.232(1) Åa | |||
| (S from thiosulfate) | 2.288(1) Åa | ||
| Cu2S Chalcocite-Q | Three, Cu–S | 2.307 Åa | 53 |
| Cu2S Chalcocite-low | Three, Cu(I)–S | 2.265 Åa | 54 |
| 2 & 3 coordinated Cu | Two, Cu(I)–S | 2.171 Åa | |
| Cu(I) in 12.88 m LiCl solution at 25 °C, 1 b | Three, Cu(I)–Cl | 2.267(7) Åb | 5 |
| Cu(I) in 15.58 m LiCl solution at 25 and 400 °C, 600 b | Three, Cu(I)–Cl | 2.272(7) Åb (25 °C) | 5 |
| Two, Cu(I)–Cl | 2.152(7) Åb (400 °C) | ||
| Cu(I) in 2 m NaHS solution at 428 °C, 600 b | Two, Cu(I)–S | 2.147(9) Åb | 42 |
| 0.4 m Cu(I) in 2 m NaCl + 0.03 m HCl solution at 325 °C (La structure) | Two, Cu(I)–Cl [CuCl2]− | 2.13 Åb | 7 |
| Cu2O, cuprite | Two, Cu(I)–O | 1.848 Åa | 55 |
| 0.4 m Cu(I) in 0.03 m HCl solution at 325 °C (Lb structure) | Two, Cu(I)–O [CuClO](aq) | 1.88 Åb | 7 |
| Gold(I) | |||
| Na3Au(S2O3)2(H2O)2 | Two, Au(I)–S (S from thiosulfate) | 2.272 Åa | 56 |
| Na3Au(S2O3)2(H2O)2 | Two, Au(I)–S (S from thiosulfate) | 2.280 Åa | 57 |
| Au2S | Two, Au(I)–S | 2.174 Åa | 58 |
| Au(I) in the H2O–S–NaOH–Na2SO4 solution at 300–450 °C, 300–600 b | Two, Au(I)–S [Au(HS)2]− | 2.29–2.30 Åb | 59 |
| Silver(I) | |||
| NaAgS2O3(H2O) | Four, Ag–S (3S + 1O) (S from thiosulfate) | 2.481 Åa | 60 |
| Ag2S Argentite (stable > 450 K) | Four, Ag–S | 2.430 Åa | 61 |
| Na3Ag(S2O3)2.(1.5)H2O | Ag1–S | 2.5994(6) Åa | 62 |
| Ag1–S | 2.5379(5) Åa | ||
| Ag1–S | 2.4384(6) Åa | ||
| Ag2–S | 2.5637(5) Åa | ||
| Ag2–S | 2.7505(6) Åa | ||
| Ag2–S | 2.4458(6) Åa | ||
| Ag3–S | 2.5518(5) Åa | ||
| Ag3–S | 2.6055(6) Åa | ||
| Ag4–S | 2.4381(6) Åa | ||
| Ag4–S | 2.7480(6) Åa | ||
| Ag4–S | 2.5396(5) Åa | ||
| Ag4–S | 2.4372(7) Åa | ||
| Three (4 times) | |||
| S from thiosulfate | |||
 | ||
| Fig. 5 UV-Visible spectra of Cu(I) chloride solutions at different salt concentrations (see Fig. S1): (a) subset I at 2 m total chloride concentration, and (b) subset II at 4 m total chloride concentration. | ||
Under our experimental conditions, a total of three chloro-complexes and three thiosulfato-complexes are expected (Table 1). Assuming a trigonal geometry for the complexes, as established on the basis of the XAS results, the possible mixed chloro-complexes include [CuCl(S2O3)(H2O)]2−, [CuCl2(S2O3)]3−, and [CuCl(S2O3)2]4−. The system is clearly underdetermined from a statistical point of view, since 7 species are sufficient to explain the whole dataset, 5 explaining 99.9% of the variance (Figure. S1b†). In addition to the large number of possible species, some of those species are expected to have similar spectra or to exist in relatively small proportion, and difficult to identify via UV-Vis spectrophotometry. Hence, it is important to use all the available chemical information in order to obtain a model that is self-consistent.
In all the fits, the formation constants of Cu(I)-chloro-complexes (Log K) in Table 1 were used. The molar absorbance spectra of these species were obtained from a fit of a series of spectra obtained under thiosulfate-free conditions (total chloride concentrations of 0.5 to 4 m) and then fixed for the rest of the fitting. As linear [CuCl(H2O)]0 is a minor species (< 10 mole%) with low molar absorption coefficients, we assumed that its absorption spectrum is equal to that of [CuCl2]−, in a manner similar to Liu et al.9 and Brugger et al.5 The spectra retrieved for linear [CuCl2]− and trigonal [CuCl3]2− are similar to those reported in ref. 5 (Fig. 6a). Slight changes in the UV-Vis spectrum above ∼4 m Cl concentrations are due to distortion of the trigonal [CuCl3]2− complex and not to a change in complex stoichiometry.5
 | ||
| Fig. 6 Results of UV-Vis data analysis for the thiosulfate-chloride dataset at 25 °C. (a) Molar absorbance spectra for individual Cu(I) complexes. (b–d) Calculated distributions of species as a function of thiosulfate concentration, for different total chloride concentrations. | ||
Similarly, the formation constant for [Cu(S2O3)2(H2O)]3− was taken from the literature (Table 1). For [Cu(S2O3)(H2O)2]−, EXAFS data reveal that it is less stable than expected from the study of Toropova et al.15 (Fig. 1a). A Log K value of 8.5 was chosen as a maximum stability for this complex consistent with our EXAFS data; this value is close to that of Golub et al.43
In the final analysis, the formation constants for [Cu(S2O3)3]5−, [CuCl(S2O3)(H2O)]2− and [Cu(S2O3)Cl2]3− were optimized. Three-dimensional residual maps show that the UV-Vis data provide only a maximum value for the formation constant of [Cu(S2O3)3]5− (Figure. S2†). The value of 9.9 for the formation of [Cu(S2O3)3]5− was chosen on the basis of the EXAFS analysis and comparison with previous studies, and corresponds to the maximum stability of this species consistent with the EXAFS data. Note that attempts to include [Cu(S2O3)2Cl]4−, the mixed ligand complex proposed by Boos and Popel,20 resulted in poor fits, and this complex is also inconsistent with the EXAFS data. Differences between the calculated and measured absorbance values are ≤ 0.10 absorbance unit. These relatively large differences (analytical reproducibility ∼0.01 absorbance unit) are attributed to the difficulty in distinguishing the spectra of the individual Cu(I) thiosulfato complexes. In the final analysis, the spectra of the three thiosulfato complexes were constrained to be equal to each other; in the absence of this constraint, fits were erratic and calculated spectra for the individual Cu(I) thiosulfato complexes were not physically realistic. Such problems are expected because of the relatively low concentrations of some of these complexes across the experimental solutions and the similarity of their molar absorbance spectra.
The final model (Table 1; Fig. 5,6) is consistent with the EXAFS results. The predicted stability of [CuCl(S2O3)(H2O)]2− is lower than that reported by Toropova et al.15 Our proposed model provides a different picture of the Cu(I) speciation in thiosulfate-rich solutions (Fig. 1b), with reduced stability of the [Cu(S2O3)(H2O)2]− and [Cu(S2O3)3]5− complexes compared to the previous consensus (Fig. 1a).
3.2 The Cu–NH3–Cl system
 | ||
| Fig. 7 XANES spectra for solutions in the Cu–NH3–Cl system, showing a smooth transition with decreasing NH3/Cu ratio (max for CuNH3_01, min for CuNH3_09). Note the three isosbestic points, labeled ‘1’ to ‘3’. | ||
Comparison of these spectra with those of Kau et al.33 indicates that the number of ligands coordinated to Cu(I) under these conditions changes from two (Sols 1 and 5, Table S3†) to three (Sol 9, Table S3†). The trigonal complex is stable at high chloride concentration, and may contain the [CuCl3]2− species described by Brugger et al.5 This hypothesis is supported by the fact that intermediary spectra (e.g., CuNH3_09) can be reproduced well by a linear combination of the spectra CuNH3_01 (mainly linear Cu(I) amine complex) and Cu_16mLiCl (mainly [CuCl3]2−,5) (Fig. 8b).
 | ||
| Fig. 8 XANES simulations of the Cu(I)–NH3–Cl system, compared with experimental data. (a) Linear end-member: solution NH3_001 (Table S3). Only raw spectra are shown. (b) Trigonal end-member: solution NH3_009 (Table S3). Fine dotted lines are convoluted spectra, all other spectra are raw calculations. | ||
As mentioned in Section 3.2.1, solution CuNH3_009 can be modeled as a linear combination of the experimental solutions CuNH3_001 (29%) and Cu_16mLiCl (71%) (Fig. 8b). Solution Cu_16mLiCl contains mainly a distorted trigonal planar [CuCl3]2− complex.5 XANES calculations for this complex5 showed that while the peak positions were modeled well, the intensities were poorly reproduced; this discrepancy was attributed to second shell effects. Fig. 8b also shows calculated spectra for the (CuN3) entity for different trigonal planar geometries, with Cu–N = 1.95 Å (Fig. 8b). The convoluted spectra are quite featureless, making them difficult to distinguish from that obtained for the (CuCl3) entity. The main difference is that the spectra for distorted trigonal configurations (T or Y shapes) show a broad peak at about 14 eV above the edge, in agreement with the experimental spectrum.

![Changes in UV-Visible spectra of Cu(i) ammonia solutions at different salt concentrations for the Cu(i)–Cl–NH3 system (see Fig. 11): (a) subset III varying Cl− concentration at ∼0.05 m ammonia and (b) subset I at [Cltot] ∼4, varying NH3 concentration. Lines are experimental data, crosses represent spectra calculated using the final fit.](/image/article/2011/RA/c1ra00708d/c1ra00708d-f10.gif) | ||
| Fig. 10 Changes in UV-Visible spectra of Cu(I) ammonia solutions at different salt concentrations for the Cu(I)–Cl–NH3 system (see Fig. 11): (a) subset III varying Cl− concentration at ∼0.05 m ammonia and (b) subset I at [Cltot] ∼4, varying NH3 concentration. Lines are experimental data, crosses represent spectra calculated using the final fit. | ||
We performed a least squares fitting of the whole dataset in order to test the nature of the ammine complexes and possible mixed chloride-ammonia species. In addition to the known copper chloride species ([CuCl(H2O)]0, [CuCl2]−, [CuCl3]2−), linear [CuNH3(H2O)]+, [Cu(NH3)2]+, and [Cu(NH3)Cl]+ complexes, the trigonal [Cu(NH3)3]+ and the trigonal mixed ammine-chloro-complexes [Cu(NH3)Cl2]− and [Cu(NH3)2Cl]− were also considered. In fitting the data, the formation constants for Cu(I)-chloro complexes were taken from Brugger et al.,5 and their molar absorbance spectra fixed from a fit of the ammonia-free solutions. Similarly, the formation constant of the [Cu(NH3)(H2O)]+ complex was taken from the literature (log β = 5.74; Table 1), since two studies provide consistent values, and this complex is expected to be present in only minor (< 10%) concentrations 44,45. In the final analysis, an excellent fit was obtained with 6 complexes [CuCl(H2O)]0, [CuCl2]−, [CuCl3]2−, [Cu(NH3)(H2O)]+, [Cu(NH3)2]+ and [Cu(NH3)Cl]0. The maximum differences in absorbance between calculated and experimental spectra were ± 0.06 absorbance units. We attribute the 7th factor in the PCA analysis (Figure S3b) to a deviation from Beer–Lambert law affecting the [CuCl3]2− species at high salinity.5 The calculated molar absorbance spectrum for [CuCl(NH3)]0 and [Cu(NH3)2]+ are consistent with linear species (Fig. 11a). The calculated distribution of species show that [Cu(NH3)2]+ is the dominant species under most conditions in ammine-bearing solutions (Fig. 11b,c); mixed chloride-amine complexes are important only under specific Cl/NH3 ratios (Fig. 11c). Fits that included mixed trigonal chloride-ammine species were statistically identical, but the molar absorbance spectra for the trigonal species were unrealistic, usually resembling the spectrum of [CuCl2]−. Also of note is that under the investigated ammonia concentrations, [Cu(NH3)3]+ is not expected to be present in significant amounts; this is consistent with the formation constant reported by previous studies (Table 1). Figure S4 plots residual contours from the fit of the data as a function of the log K for the formation of the linear diamminecopper(I) and amminechlorocopper(I) complexes. A well-constrained 90% confidence interval gives an estimate of the Log Ks for [Cu(NH3)2]+ = 10.7(+ 0.2/−0.5) and [CuClNH3]0 = 8.38(+ 0.2/−0.3) at 25 °C.
 | ||
| Fig. 11 Results of UV-Vis data analysis for the NH3-chloride dataset at 25 °C. (a) Molar absorbance spectra for individual Cu(I) complexes. Note that the spectrum of Cu(NH3)3+ could not be retrieved because it is a minor component in the investigated solutions. (b,c) Calculated distributions of species as a function of NH3 concentration for 0.0 and 4.0 m NaCl solutions. | ||
4. Conclusions
By combining UV-Vis and XAS data, this study has provided a speciation model for copper(I) in mixed thiosulfate-chloride and ammonia-chloride systems consistent with UV-Vis and XAS data (Fig. 1). The new formation constants derived in our study allow to refine the prediction of Cu(I) speciation in ammonia-chloride solutions. For the thiosulfate-chloride system, our results indicate that the [Cu(S2O3)2(H2O)]3− complex is more stable than previously thought. We demonstrate the existence of mixed thiosulfate-chloride complexes ([Cu(S2O3)Cl(H2O)]2− and [Cu(S2O3)Cl2]3−), with the former complex being dominant under some conditions (Fig. 1b). The results of our study allow more comprehensive predictions of speciation which can be applied in the design of efficient hydrometallurgical gold processing methods using thiosulfate lixiviants, including where using chloride-rich waters commonly encountered in gold deposits in semi-arid environments.This study illustrates the complexity of studying speciation in apparently ‘simple’ inorganic systems. This complexity is related to the fact that many species can co-exist in solution; that is, in many cases individual species cannot be isolated due to the presence of significant amounts of other species (see speciation diagrams in Fig. 6b–d and 10b,c), which may be only minor components (≤ 15%). As a consequence, in order to study these complex systems effectively, multiple experimental methods, such as XAS and UV-Visible spectrophotometry, are necessary.
Our approach emphasizes the importance of understanding the coordination geometry when studying the speciation in such systems. XANES spectroscopy in particular is a powerful tool to establish the geometry of the dominant complexes in solution, thereby constraining the number of possible species that may exist in solution. Information about the coordination geometry of the complexes is also important in assessing their catalytic potential, or in comparing results from theoretical molecular dynamic studies with experimental results (e.g., see ref. 12).
Acknowledgements
This work was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program and the Australian Research Council through the Centre for Green Chemistry. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Contract No.W-31-109-Eng-38. JB acknowledges an Australian Research Council Professorial Fellowship.References
- G. J. Sparrow and J. T. Woodcock, Extractive Metall. Rev., 1995, 14, 193–247 CrossRef.
- M. G. Aylmore and D. M. Muir, Miner. Eng., 2001, 14, 135–174 CrossRef CAS.
- M. G. Aylmore and D. M. Muir, Miner. Metall. Proc., 2001, 18, 221–227 CAS.
- Water Management at KCGM. URL: http://www.superpit.com.au/Environment/Water/tabid/129/Default.aspx..
- J. Brugger, B. Etschmann, W. Liu, D. Testemale, J.-L. Hazemann, H. Emerich, W. van Beek and O. Proux, Geochim. Cosmochim. Acta, 2007, 71, 4920–4941 CrossRef CAS.
- L. Ciavatta and M. Iuliano, Ann. Chim., 1998, 88, 71–89 CAS.
- J. L. Fulton, M. M. Hoffmann and J. G. Darab, Chem. Phys. Lett., 2000, 330, 300–308 CrossRef CAS.
- W. H. Liu, J. Brugger, B. Etschmann, D. Testemale and J. L. Hazemann, Geochim. Cosmochim. Acta, 2008, 72, 4094–4106 CrossRef CAS.
- W. Liu, J. Brugger, D. C. McPhail and L. Spiccia, Geochim. Cosmochim. Acta, 2002, 66, 3615–3633 CrossRef CAS.
- W. Liu, D. C. McPhail and J. Brugger, Geochim. Cosmochim. Acta, 2001, 65, 2937–2948 CrossRef CAS.
- Z. Xiao, C. Gammons and A. Williams-Jones, Geochim. Cosmochim. Acta, 1998, 62, 2949–2964 CrossRef CAS.
- D. M. Sherman, Geochim. Cosmochim. Acta, 2007, 71, 714–722 CrossRef CAS.
- R. M. Smith and A. E. Martell, Critical Stability Constants, Vol. 4: Inorganic Complexes, Plenum Press, 1976 Search PubMed.
- A. M. Golub, S. S. Butsko and L. P. Dobryanskaya, Z. Neorganich. Khim., 1975, 20, 2728–2732 CAS.
- V. F. Toropova, I. Sirotina and T. Lisova, Uchenye Zapiski Kazanskogo Gosudarst. Univ., 1955, 115, 43–52 CAS.
- A. E. Martell, R. J. Motekaitis, Determination and Use of Stability Constants, VCH Publishers, Inc., 1988 Search PubMed.
- A. I. Stabrovskii, Z. Obshch. Khim., 1951, 21, 1223–1237 Search PubMed.
- E. A. Ukshe and A. I. Levin, Z. Fizich. Khim., 1953, 27, 1396–1403 CAS.
- D. G. Davis, Anal. Chem., 1958, 30, 1729–1732 CrossRef CAS.
- G. A. Boos and A. A. Popel, Russian J. Inorg. Chem., 1970, 15, 792–793 Search PubMed.
- J. S. Solis, G. Hefter and P. M. May, Aust. J. Chem., 1995, 48, 1293–1300 CrossRef.
- A. I. Vogel, Vogel's Textbook of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis, Longman Inc., 1978 Search PubMed.
- L. N. Var'yash, Geochem. Int., 1989, 26, 80–90 Search PubMed.
- J. Brugger, D. C. McPhail, J. Black and L. Spiccia, Geochim. Cosmochim. Acta, 2001, 65, 2691–2708 CrossRef CAS.
- J. Brugger, Comput. Geosci., 2007, 33, 248–261 CrossRef.
- H. C. Helgeson, Am. J. Sci., 1969, 267, 729–804 CrossRef CAS.
- H. Helgeson and D. H. Kirkham, Am. J. Sci., 1974, 274, 1089–1198 CrossRef CAS.
- J. Kielland, J. Am. Chem. Soc., 1937, 59, 1675–1678 CrossRef CAS.
- H. C. Helgeson, D. H. Kirkham and G. C. Flowers, Am. J. Sci., 1981, 281, 1249–1516 CrossRef CAS.
- S. M. Heald, D. L. Brewe, E. A. Stern, K. H. Kim, D. T. Jiang, E. D. Crozier and R. A. Gordon, J. Synchrotron Radiat., 1999, 6, 347–349 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- J. Chaboy, A. Munoz-Paez, P. J. Merkling and E. S. Marcos, J. Chem. Phys., 2006, 124, 064509-1–064509-9 CrossRef.
- L.-S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- Y. Joly, Phys. Rev., 2001, 125120-1–125120-10 Search PubMed.
- D. Testemale, J. Brugger, W. Liu, B. Etschmann and J.-L. Hazemann, Chem. Geol., 2009, 264, 295–310 CrossRef CAS.
- M. O. Krause and J. H. Oliver, J. Phys. Chem. Ref. Data, 1979, 8, 329 CrossRef CAS.
- J. D. Bourke, C. T. Chantler and C. Witte, Phys. Lett. A, 2006, A360, 702–706 Search PubMed.
- Y. Joly, D. Cabaret, H. Renevier and C. R. Natoli, Phys. Rev. Lett., 1999, 82, 2398–2401 CrossRef CAS.
- D. Testemale, J. L. Hazemann, G. S. Pokrovski, Y. Joly, J. Roux, R. Argoud and O. Geaymond, J. Chem. Phys., 2004, 121, 8973–8982 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B, 1998, 58, 7565–7576 CrossRef CAS.
- C. H. Booth and F. Bridges, Physica Scripta, 2005, 2005, 202–204 CrossRef.
- B. E. Etschmann, W. Liu, D. Testemale, H. Müller, N. A. Rae, O. Proux, J. L. Hazemann and J. Brugger, Geochim. Cosmochim. Acta, 2010, 74, 4723–4739 CrossRef CAS.
- A. M. Golub, L. P. Dobryanskaya and S. S. Butsko, Z. Neorganich. Khim., 1976, 21, 2733–2737 CAS.
- J. Bjerrum, Kgl. Danske Videnskab. Selskab, Math.-fys. Medd., 1934, 12, 67pp Search PubMed.
- T. V. Stupko, V. E. Minorov and G. L. Pashkov, Z. Priklad. Khim., 1998, 71, 1087–1090 CAS.
- S. Lurdes, S. Gonçalves and L. Sigg, Electroanalysis, 1991, 3, 553–557 CrossRef.
- J. Bjerrum, Acta Chem. Scand., 1986, A40, 223–235 Search PubMed.
- O. Horvath and K. L. Stevenson, Inorg. Chem., 1989, 28, 2548–2551 CrossRef CAS.
- S. B. Black, The Thermodynamic Chemistry of the Aqueous Copper-Ammonia Thiosulfate System, PhD thesis, Murdoch University, 2006, 261pp Search PubMed.
- A. Ferrari, A. Braibanti and A. Tiripicchio, Acta Crystallogr., 1966, 21, 605 CrossRef CAS.
- R. Ramirez, C. Mujica, A. Buljan and J. Llanos, Bol. Soc. Chilena De Quimica, 2001, 46, 235–245 CAS.
- J. C. Monier and R. Kern, Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences, 1955, 241, 69–71 CAS.
- A. Janosi, Acta Crystallogr., 1964, 17, 311–312 CrossRef CAS.
- H. T. Evans, Nature Physical Science, 1971, 232, 69–70 CAS.
- A. Kirfel and K. Eichhorn, Acta Cryst. A, 1990, 46, 271–284 CrossRef.
- H. Ruben, A. Zalkin, M. O. Faltens and D. H. Templeton, Inorg. Chem., 1974, 13, 1836–1839 CrossRef CAS.
- R. F. Baggio and S. Baggio, J. Inorg. Nucl. Chem., 1973, 35, 3191–3200 CrossRef CAS.
- K. Ishikawa, T. Isonaga, S. Wakita and Y. Suzuki, Solid State Ionics, 1995, 79, 60–66 CrossRef CAS.
- G. S. Pokrovski, B. R. Tagirov, J. Schott, E. F. Bazarkina, J. L. Hazermann and O. Proux, Chem. Geol., 2009, 259, 17–29 CrossRef CAS.
- L. Cavalca, A. Mangia, C. Palmieri and G. Pelizzi, Inorg. Chim. Acta, 1970, 4, 299–304 CrossRef CAS.
- R. J. Cava, F. Reidinger and B. J. Wuensch, J. Solid State Chem., 1980, 31, 69–80 CrossRef CAS.
- E. J. Chan, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2008, 634, 2825–2844 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: composition of solutions for XAS and UV-Visible measurements (Tables S1–S4); results of principle component analysis and residual maps for the thiosulfate-chloride and ammonia-chloride data UV-Visible data (Figures S1–S4). See DOI: 10.1039/c1ra00708d/ |
| This journal is © The Royal Society of Chemistry 2011 |