The effect of temperature on the internal dynamics of dansylated POPAM dendrimers†
Jukka
Aumanen
*a,
Gilberto
Teobaldi
b,
Francesco
Zerbetto
c and
Jouko
Korppi-Tommola
a
aDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FIN-40014, Jyväskylä, Finland. E-mail: jukka.aumanen@jyu.fi
bStephenson Institute for Renewable Energy and Surface Science Research Centre, Department of Chemistry, The University of Liverpool, Liverpool, L69 3BX, United Kingdom
cDipartimento di Chimica “G. Ciamician”, Università di Bologna, via F. Selmi 2, 40126, Bologna, Italy
First published on 27th October 2011
Abstract
The internal and rotational dynamics of the dansylated poly(propylene amine) dendrimers (POPAM) have been studied by time correlated single photon counting (TCSPC) and molecular dynamics (MD) simulations. The hydrodynamic volumes of the dendrimer generations from G1 to G4 were estimated by fluorescence anisotropy data. Experiments and simulations suggest that the volume and the shape of the dendrimers are temperature dependent. At low temperatures the dendrimer structure becomes more spacious and rigid and back-folding of the individual branches is slowed down. For the G3 and G4 generations the temperature effects are much stronger than for the smaller G1 and G2 generations, where back-folding does not play a significant role. MD simulations elucidate the temperature-driven contraction, which is governed by the balance between intra-dendrimer and short-range solvent–dendrimer interactions and is further tuned by the dependence on the dendrimer generation, functionalization, and solvent. These findings pave the way to the design of dendrimers with temperature-dependent volume, accessible area, and host–guest chemistry.
Introduction
Symmetrically branched dendrimers have attracted scientists for over a quarter of a century.1,2 At first, the focus of the attention was the unique structure of dendrimers and the synthetic challenges associated to their production.3,4 Later on, the fundamental properties and possible applications of dendrimers became central and were extensively studied.5–7 The capability of dendrimers to encapsulate smaller guests molecules, such as drugs, catalysts, and chromophores is an especially interesting property since it opens the way to possible functional applications.8,9 For this reason, the size and the shape of the dendrimers in solution are considered of particular interest as they affect the encapsulation properties of the dendrimers. Time resolved spectroscopic measurements can be used to investigate the internal dynamics and structural properties of dendrimers labelled with fluorescent chromophores.10–13‘Soft’ dendrimers continuously change their shape as a result of back-folding of the dendritic arms. Dendrimers may be assumed to attain a ‘time averaged’ spherical shape since the motions of the dendritic arms occur on a faster time scale than the overall molecular rotation. As the generation number grows, dendrimers tend to be more spherical.14,15 The motions of dendrimers can be divided into three categories: (1) molecular rotational motion (2) bending of dendritic branches and (3) local motions of the terminal groups.16 In dendrimers with peripheral probe chromophores, experimentally observed dynamics are an ensemble average of these motions. In larger dendrimers, back-folding motions complicate the overall dynamic picture further. Dendrimer branches that are oriented away from the core move quite freely, while the motions of the back-folded branches are hindered by the dendrimer backbone. The flexibility of the dendrimers allows manipulation of their size and shape in solution by varying the solvent or pH17–19, and the temperature20 has also been predicted to affect to the size of the dendrimers.
Poly(propylene amine) dendrimers (POPAM) can be functionalized with dansyl chromophores to make them absorb UV light and emit visible light.21 Dansylated POPAM dendrimers are capable of encapsulating xanthene dyes to form host–guest complexes.22 In our previous studies, we have reported the rates of energy transfer from dansyls of three POPAM dendrimers to their eosin guests.23 We also discussed the internal dynamics of the dendrimers in solution.10 Transient absorption and streak-camera methods allowed resolving kinetic constants up to about one nanosecond. In this work, we extend the measurements to lower temperatures and to longer time scales by using a time correlated single photon counting (TCSPC) method. The time scale of the method allows the entire decay of dansyl fluorescence to be seen and the rotational correlation times of the G3 and G4 dendrimers, which fall in the nanosecond domain, to be resolved. The fluorescence anisotropies were recorded to obtain information on the internal dynamics and volumes of the dendrimers at low temperatures. Molecular dynamics simulations for the G4 dendrimer were performed to give background for the interpretation of the experimentally recorded dynamics and to provide atomistic insight into the size and shape of the dendrimer as a function of temperature.
Both experiments and theory indicate a profound effect of the temperature on the structure of the dendrimer and, consequently, on its internal and global dynamics. Rather unexpectedly, we find that the size of the larger-generation dendrimers (G3–G4) decrease as the temperature is increased. Since altering the structure of the dendrimer may be advantageous for a controlled release of the guest, our findings introduce new possibilities in the host–guest applications of POPAM dendrimers. On this basis, fluorescence anisotropy measurements for dendrimer–eosin complexes were also carried out in order to gain insight into the behaviour of the guest molecule at lowered temperatures.
Experimental
The synthesis of the dansylated dendrimers is described elsewhere.21,24 Structures of the investigated molecules are presented in Fig. 1. Dendrimer samples were dissolved in chloroform and the optical density of each sample at the excitation wavelength, 375 nm, was tuned to between 0.1 and 0.2 in a 1 cm path length cuvette. To form complexes, a droplet of highly concentrated eosin in ethanol solution was added to the dendrimer solutions and mixed vigorously. Samples were then diluted with the dendrimer solution to get an optical density (OD) of between 0.1 and 0.2 at the eosin excitation wavelength, 485 nm. Dendrimer eosin complexes had then approximately one eosin molecule per eight dansyl units, corresponding to average of one eosin per G2, two eosins per G3 and four eosins per G4 molecule.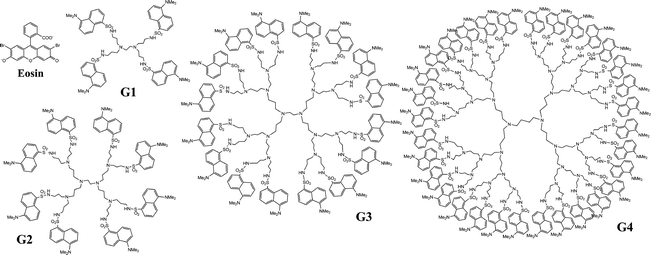 | ||
| Fig. 1 Schematic structures of the dansylated POPAM dendrimers and the eosin dye. | ||
Time resolved fluorescence measurements were performed with a commercial PicoQuant HydraHarp 400 time correlated single photon counting data acquisition system. The excitation source comprised of a PicoQuant PDL 800-D pulsed diode laser driver with 375 nm (spectral FWHM 1.3 nm) and 485 nm (4.0 nm) laser heads. The repetition rate of the excitation pulses was set to 10 MHz in all measurements and the output power of the lasers were 0.74 mW and 0.60 mW for 375 and 485 nm excitations, respectively. The time resolution of the experiment was determined to be 100 ps for a 375 nm laser and 180 ps for a 485 nm laser. To collect fluorescence light around the emission maxima of dansyls and eosin, interference filters with centre wavelengths of 499 nm (spectral bandwidth 10.6 nm) and 558 nm (11.8 nm), respectively were used. It is noted that the wide fluorescence spectrum of the dansyl partially spans the spectrum of the eosin and the filter does not remove dansyl emission completely. Signals with parallel and perpendicular polarizations were measured by adjusting the polarization of the excitation pulses with a Berek polarization compensator (New Focus). The excitation beam was guided through a Glan laser polarizer to the sample and a film polarizer was installed in front of a MCP-PMT detector. As the orientation of the detected polarization was kept constant, no correction for the detector response with respect to parallel and perpendicular signals was needed. The input rate of the pulses falling on the detector was kept lower than one hundredth of the repetition rate of the excitation laser by controlling the excitation energy with a variable neutral density filter. Under experimental conditions, the energy of the excitation pulses on the sample was from one to four picojoules per pulse. The excitation energy was kept constant for parallel and perpendicular polarizations. Liquid state samples were cooled in a liquid nitrogen cooled cryostat (Oxford Optistat DN) containing a heater for temperature control. Sample temperatures were always kept above the freezing point of the solvent (−63.5 °C). All experiments were carried out for air-equilibrated solutions at ambient atmosphere.
Simulations
Molecular dynamics (MD) simulations were carried out via a modified Beeman algorithm as implemented in the TINKER program.25 The same approach was previously used to study eosin (E) and Rose Bengal (RB) encapsulation and dynamics in dansylated POPAM dendrimers.10,26–28 The adopted force-field was MM3.29 Given the MM3 approximation to the electrostatic interactions on the basis of bond-centred dipoles alone,29 and following previous successful applications of the method,10,26–28 we relied on the TINKER defaults for the evaluation of the MM3 electrostatic interactions using a cutoff value of 10 Å. As for the van der Waals interactions, in this case we also relied on the TINKER default by adopting a 9 Å cutoff value. The 2 ns MD production runs of the POPAM G4 dendrimer in CHCl3 (1728 molecule) were generated by sampling in the canonical ensemble (NVT) at T = 213, 233, 253, 273 K. In view of the rotational correlation analysis (see below) the MD trajectories were extended by a further 8 ns, accounting for a total simulation time of 10 ns (see also the ESI†). Following previous successful applications of the method,10,26–28 in all the simulations we used the Berendsen thermostat,30 as implemented in the TINKER program.25 For each NVT simulation, the side of the cubic simulation cell was obtained following 1 ns NPT equilibration (P = 1 atm) at the considered temperature. The equilibrated values go from 62.824 Å (213 K) to 64.691 Å (273 K). In all of the simulations, the integration time step was 1 fs and no geometrical constrain was enforced with respect to the hydrogen atoms. Due to the known tendency of the adopted force-field to underestimate (by ∼25%) the CHCl3 density and inter-molecular interactions31, it was not possible to obtain a converged NPT equilibration at 293 K. As a result, this temperature was not considered further in the theoretical study.The excluded volume was calculated using Kundrot et al.'s modified version32 of Connolly's original analytical description of molecular surfaces,33–34 as implemented in the TINKER program.25 In all cases, the G4 excluded volume was calculated adopting a 2 Å -radius for the atomic probe.
The nine components of the 3 × 3 dendrimer inertia tensor (I)
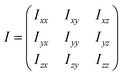 | (1) |
were calculated as:35
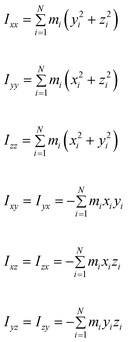 | (2) |
where N is the total number of the dendrimer atoms and mi refers to the mass of the ith atom. The G4 principal inertia moments (PIMs) were obtained by diagonalizing I with the method of Jacobi rotation,36 as implemented in the TINKER program.25
Following Ref. 11, the global dendrimer rotational autocorrelation function (RACF) was calculated on the basis of extended 10 ns MD trajectories as:
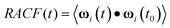 | (3) |
where ωi(t) defines the set of the three orientation cosines which the ith atom forms with the three axes of the reference system (centred on the instantaneous centre of mass of the dendrimer):
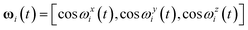 | (4) |
The RACF was calculated using blocks of 2500 points (2.5 ns) and eventually averaged over 7500 (t0) origins. The calculated RACF(t) was well fitted by a single-exponential:
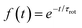 | (5) |
yielding the slow rotational decay rate, τrot.
Finally, following Ref. 37, the temperature-dependent CHCl3 liquid-phase entropy, Ssolv, was calculated as:
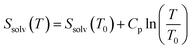 | (6) |
using the experimental values of 295.51 J mol−1 K−1 and 114.25 J mol−1 K−1 for the CHCl3 molar entropy at 298 K [Ssolv(T0 = 298 K)]38 and constant-pressure specific heat (Cp),39 respectively. By assuming the CHCl3 molecules frozen inside the dendrimer [hence with zero entropy, Ssolv@G4(T) = 0], it is possible to approximate (overestimate) the change in entropy ΔSsolv(T) due to the molecules leaving the dendrimer for the bulk-solvent region by Ssolv(T) alone [ΔSsolv(T) = Ssolv(T) − Ssolv@G4(T) ∼ Ssolv(T)].
Results
Dansylated POPAMs
Magic angle fluorescence decay was constructed from measured parallel and perpendicular traces by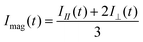 | (7) |
Lowering the temperature from 293 K to 213 K lengthened the fluorescence lifetime from ca. 13 ns to 16 ns without significant differences between the dendrimer generations. Single exponential fluorescence lifetimes of the G1 to G4 dendrimers are presented in Table 1.
| Emission | Anisotropy decay | |||
|---|---|---|---|---|
| T/K | lifetime (ns) | τ1 (ns) | τ2 (ns) | |
| G1 | 213 | 16.0 | 1.58 (100%) | |
| 233 | 15.3 | 1.00 (100%) | ||
| 253 | 14.5 | 0.63 (100%) | ||
| 273 | 13.9 | 0.42 (100%) | ||
| 293 | 13.6 | 0.30 (100%) | ||
| G2 | 213 | 15.8 | 0.61 (24%) | 4.29 (76%) |
| 233 | 15.1 | 0.45 (25%) | 2.47 (75%) | |
| 253 | 14.5 | 0.37 (31%) | 1.63 (69%) | |
| 273 | 13.8 | 0.36 (43%) | 1.21 (57%) | |
| 293 | 13.6 | 0.42 (72%) | 1.17 (28%) | |
| G3 | 213 | 16.0 | 0.85 (21%) | 9.24 (79%) |
| 233 | 15.3 | 0.56 (23%) | 5.11 (77%) | |
| 253 | 14.7 | 0.46 (34%) | 3.17 (66%) | |
| 273 | 14.0 | 0.35 (33%) | 2.05 (67%) | |
| 293 | 13.7 | 0.34 (44%) | 1.48 (56%) | |
| G4 | 213 | 15.9 | 2.09 (23%) | 19.1 (77%) |
| 233 | 15.2 | 1.46 (28%) | 11.1(72%) | |
| 253 | 14.5 | 0.94 (33%) | 6.54 (67%) | |
| 273 | 13.7 | 0.54 (36%) | 4.02 (64%) | |
| 293 | 13.2 | 0.40 (44%) | 2.61 (56%) | |
Fluorescence anisotropy decays were calculated as
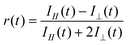 | (8) |
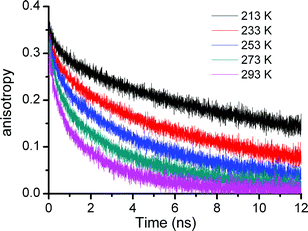
The anisotropy signals showed a clear dependence on the dendrimer generation and temperature. Anisotropy decay curves for the G4 POPAM dendrimer at different temperatures are shown in Fig. 2. Fig. 3 presents the anisotropy decay traces for the G1 to G4 dendrimers at room temperature (293 K) and at 213 K. The corresponding time constants from two exponential fits (except monoexponential for G1) are collected in Table 1. For the G2, G3 and G4 dendrimers, both the fast and the slow decay components became longer and the relative amplitude of the longer component grew as the temperature was lowered. For the monodansyl reference compound, N-propyldansylamine, anisotropy decay could not be resolved at room temperature, since it was determined to be faster than the time resolution of the present instrumentation.10
 | ||
| Fig. 2 Fluorescence anisotropy decay of the G4 dendrimer in chloroform at temperatures between 213 K and 293 K. | ||
 | ||
| Fig. 3 Fluorescence anisotropy decay of the Gn (n = 1–4) dendrimers and N-propyldansylamine reference compound (N) at (a) room temperature (293 K) and (b) 213 K. The filled area on the left shows the time resolution of the experiment. | ||
POPAM–eosin complexes
Excitation of the dansyls of the dendrimer–eosin complexes resulted in both dansyl and eosin fluorescence. Dansyl fluorescence was quenched with respect to that of the pristine dendrimers, but the fluorescence lifetimes of the complexes were very close to the lifetimes of the pristine dendrimers and practically independent of the dendrimer generation (Table 2). The lifetime of the eosin fluorescence was shorter than that of the eosin in solution and it became shorter as the size of the host dendrimer grew. However, it was rather insensitive to temperature changes (Table 2). The fluorescence lifetime of eosin in ethanol solution was 3.6 ns and did not depend on temperature.Eosin fluorescence anisotropy could not be observed after dansyl excitation due to the random relative orientations of the donor and the acceptor. When eosins were excited directly, the initial anisotropy for the G2 complex was close to 0.4, whereas for the G3 and G4 complexes the initial anisotropies were 0.15 and less than 0.1, respectively. The eosin fluorescence anisotropy decay rates of the complexes were close to the values observed for the dansyl fluorescence anisotropies of corresponding pristine dendrimer generations; it was fastest in G2 complexes and slowest in G4 complexes.
Simulation of the G4 POPAM dendrimer
Molecular dynamics simulations at different temperatures offer insight into the temperature effects on the structure of the G4 POPAM dendrimer.Starting with the G4 global structure, and assuming a spherical shape for the dendrimer, the simulations suggest a progressive reduction of the time-averaged gyration radius (rgyr)26 of G4 from 15.3 Å to 14.0 Å as the simulated temperature increases from 213 K to 273 K (Fig. 4).
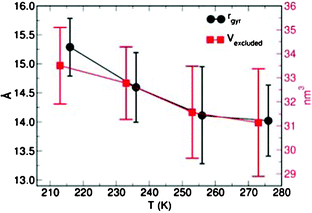 | ||
| Fig. 4 Time-averaged gyration radius (rgyr) and excluded volume (Vexcluded, nm3) for G4 at several temperatures. For clarity, the rgyr dataset has been shifted by +5 K. In both panels the error bars indicate the maximum deviations of the instantaneous value from the average. | ||
The definition of the gyration radius relies on the assumption that G4 is spherical,26 an approximation which may not always be accurate for POPAM dendrimers (see below). The rgyr results were checked against the time-average of the calculated principal inertia moments (PIMs). As shown in Table 3, the temperature-driven contraction of the PIMs confirms that the reduction of the G4-size with increasing temperature is representative.
| T/K | I x | I y | I z | I y /Iz | I x /Iy | I x /Iz | a |
|---|---|---|---|---|---|---|---|
| 213 | 1.425 (0.186) | 1.856 (0.242) | 2.282 (0.280) | 0.813 | 0.768 | 0.624 | 0.018 |
| 233 | 1.393 (0.164) | 1.741 (0.200) | 1.923 (0.284) | 0.905 | 0.800 | 0.724 | 0.009 |
| 253 | 1.252 (0.145) | 1.674 (0.300) | 1.836 (0.256) | 0.911 | 0.748 | 0.681 | 0.012 |
| 273 | 1.286 (0.192) | 1.502 (0.169) | 1.906 (0.269) | 0.788 | 0.856 | 0.674 | 0.013 |
To further investigate this phenomenon, we also calculated the G4 excluded volume (Vexcluded) as a function of the temperature. These results are displayed in Fig. 4. In line with both the rgyr and PIMs analyses, the simulations also indicate a decrease in the G4-volume as the temperature is increased.
Here we note that the calculated G4 contraction at high temperature starkly deviates from the temperature-driven volume increase predicted by Flory-type models on starburst dendrimers.40 Thus, explicit modelling with atomistic potentials of the solvent turns out to be very important for representative simulation of POPAM dendrimers in CHCl3 and, we speculate, in other organic solvents.
While useful for the description of the G4 structure and shape, the time-averaged rgyr, PIMs and Vexcluded do not offer any information about the distribution of the dansyl units in the G4 dendrimer. Such information is provided in Fig. 5a as a histogram of the distances of the dansyls from the G4 centre of mass (CM). Consistent with the reduction of the G4 volume, the dansyl distribution peak progressively shifts closer to the dendrimer CM as the temperature is increased from 213 K to 273 K.
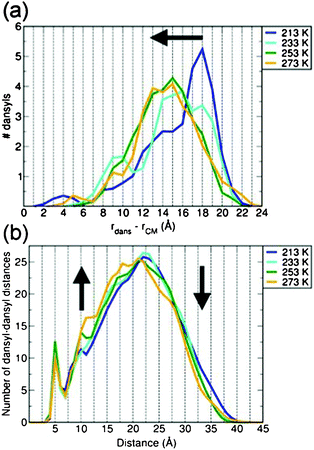 | ||
| Fig. 5 (a) Time-averaged occurrence histogram of the distances between the dansyls and centre of mass (CM) of G4 at several temperatures. (b) Time-averaged dansyl–dansyl distance occurrence histograms at the same temperatures. The temperature-induced shifts in the calculated distributions have been highlighted by black arrows. All distributions have been generated on the basis of 1 Å sampling grids, and normalized to the total number of dansyl fragments (32) and dansyl-dansyl distances (496), in panel (a) and (b), respectively. | ||
The dependence of the G4 structure on the temperatures also affects the distribution of the distances between the dansyls. Unsurprisingly, as the temperature is increased, the reduction of the G4-size is accompanied by a parallel change in the dansyl–dansyl distance distribution, leading to larger and smaller occurrences for medium- (10–20 Å) and long-range (>20 Å) separations, respectively (Fig. 5b).
The temperature-driven change in the extension and shape of G4 also affects the interactions between the dendrimer and solvent molecules. Fig. 6 reports the time-averaged number of solvent molecules within rgyr, and in close-contact with G4. Following Ref. 10, 26, close-contact solvent molecules were identified on the basis of a 2.8 Å cutoff value. The calculated changes in rgyr (Fig. 4), leading to a more compact G4 structure, are accompanied by a parallel decrease of solvent molecules within rgyr. Notably, the number of solvent molecules in close-contact with G4 also decrease as the temperature is increased from 213 K to 273 K. This result originates from the dendrimer contraction at higher temperatures and the ensuing reduction of the surface accessible to the solvent, which in turn leads to fewer CHCl3 molecules in (close-) contact with G4.
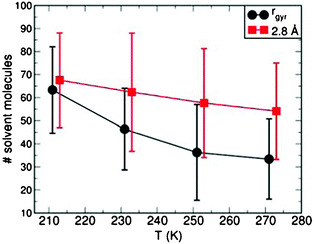 | ||
| Fig. 6 The average number of CHCl3 molecules within the G4 gyration radius (rgyr), and less than 2.8 Å away from G4 as a function of the simulated temperature. For clarity, the close-contact solvent dataset (2.8 Å) has been shifted by +5 K. The error bars indicate the maximum deviations of the instantaneous values from the corresponding average. | ||
The energy factors driving the G4 contraction at higher temperature can be identified by the analysis of the different contributions to the total energy of the system. As shown in Fig. 7a, the increased thermal energy at high temperatures allows the population of high-energy configurations for both G4 and the solvent (CHCl3). The higher energy of these configurations stems from several factors, namely (i) the mechanical (mainly torsional) energy penalization of the back-folded G4, (ii) the reduction of the solvent–solvent interactions due to the lower CHCl3 density at high temperatures, and (iii) the smaller G4–CHCl3 interactions due to the reduction of the solvent-accessible area (Fig. 6).
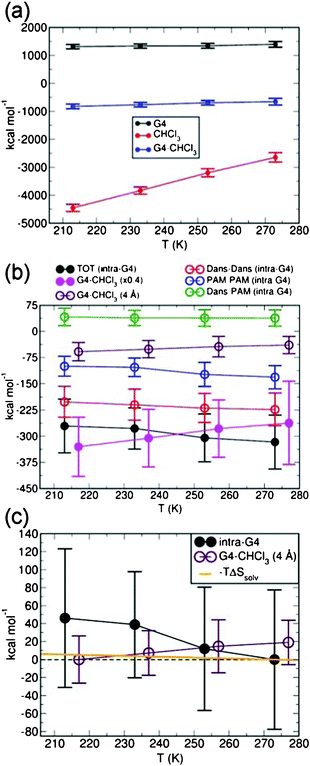 | ||
| Fig. 7 (a) Time-averaged dendrimer (G4), solvent (CHCl3) and G4–CHCl3 interaction energy at the considered temperatures. (b) Comparison between the total intra-dendrimer (intra-G4) and G4–CHCl3 interaction energy (filled circles). Note that, for clarity, the G4–CHCl3 interaction energy has been multiplied by a factor of 0.4. The dansyl (Dans)–Dans, propylene amine (PAM)–PAM and Dans–PAM contributions (empty circles) to the total (TOT) intra-G4 energy are also reported. The contribution to the total G4–CHCl3 interaction energy from the solvent molecules less than 4 Å away from G4 is also shown by empty circles and labelled. (c) Comparison between the total intra-G4 energy, the contribution from solvent molecules less than 4 Å away from G4, and the entropy gain (−TΔSsolv) due to the CHCl3 molecules leaving the dendrimer for the bulk-solvent phase. For ease of comparison between the trends, the intra-G4 and G4–CHCl3 datasets have been aligned by subtracting the corresponding minimum value. The entropy gain (−TΔSsolv) has been scaled with respect to the 273 K value. In all panels, the error bars indicate the maximum deviations of the instantaneous values from the corresponding average. For clarity, the G4–CHCl3 datasets in panels (b–c) have been shifted by +10 K. | ||
Comparison between the G4–CHCl3 and the intra-dendrimer (intra-G4) interactions (Fig. 7b) indicates that the reduction of G4–CHCl3 interactions at high temperature is accompanied by a parallel increase in the intra-G4 interactions. The intra-G4 energy gain is due to larger dansyl–dansyl and propylene amine (PAM)–PAM interactions which (over-) compensate for the dansyl-PAM’s (slight) repulsion in the back-folded G4.
Consistent with G4 solubility in CHCl3, the G4–CHCl3 energy is significantly larger (∼2.5 times) than the intra-G4 interaction at all the considered temperatures. Here we note that the G4–CHCl3 interaction contains several contributions due to (i) the solvent molecules inside the G4, (ii) the solvent molecule in close-contact with G4 or equivalently in its 1st solvation sphere, and (iii) the interactions between G4 and the outer solvation spheres. To jointly analyse the contributions due to the 1st solvation sphere and the CHCl3 molecules inside G4, we adopted a 4 Å cutoff to isolate the G4 interactions with close-contact solvent molecules. As shown in Fig. 7b, the results of this analysis indicate that most of the G4–CHCl3 interaction energy is due to long-range interactions between G4 and the outer solvation sphere rather than to close-contact solvent molecules.
Crucially, the energy gain due to intra-G4 contributions (∼45 kcal mol−1, Fig. 7c) between 213 K and 273 K is roughly three times the energy penalization due to the reduced close-contact G4–CHCl3 interactions (∼16 kcal mol−1). Thus, as the increased temperature populates higher energy back-folded configurations, it is energetically convenient to maximize the intra-dendrimer interactions by removing close-contact solvent molecules.
For completeness, we also assessed the importance of the entropic gain due to the solvent molecules leaving the dendrimer for the bulk-solvent region. By assuming that the CHCl3 molecules are frozen inside the dendrimer (hence without any entropy, clearly an underestimation of the solvent–entropy inside G4), it is possible to estimate the entropy gain by simply calculating the solvent phase CHCl3 entropy at the simulated temperatures (eqn (6)). The results of this analysis, displayed in Fig. 7c, indicate that the entropic increase between 213 K and 273 K due to the CHCl3 molecules leaving the dendrimer is roughly 6 kcal mol−1. This value is too small to counterbalance the loss in dendrimer–solvent interactions (∼16 kcal mol−1) and more than seven times smaller than the intra-G4 stabilisation (∼45 kcal mol−1). Due to the adopted approximation of frozen CHCl3 molecules within G4, we expect the actual entropic gain to be even smaller.
On these grounds, the G4 contraction can be rationalized as due to the maximization of the more favourable intra-G4 interactions and to the ensuing stabilization of high-energy back-folded configurations, with marginal contributions from the entropic gain due to the solvent molecules leaving the dendrimer.
Finally, in an attempt to make semi-quantitative contact with the rotational component of the experimentally measured anisotropy decays rates (τ2 in Table 1), we also estimated the effect of the temperature-driven changes in the dendrimer size and interactions on the G4 rotational correlation time. Table 4 reports the G4 dendrimer correlation times τrot as calculated from the computed rotational autocorrelation function (RACF) at several temperatures. Consistent with the increased thermal energy and the calculated decrease of both G4-size and solvent density, τrot decreases as the temperature is increased from 213 K to 273 K. The calculated values can be rather accurately (max error ∼ 5%) fitted to an exponential decay ln(τrot) = ln(τ0) − βT with β = 0.024 K−1.
| T/K | τ rot (ns) | |
|---|---|---|
| G4 | 213 | 6.86 |
| 233 | 3.80 | |
| 253 | 2.60 | |
| 273 | 1.55 |
Discussion
Fluorescence lifetimes and anisotropy decays
The absorption and emission spectra, lifetimes and quantum yields of dansylated POPAMs are almost identical to the corresponding properties of the monodansyl compound.41 The observed independency of the fluorescence lifetime on the dendrimer generation suggests only weak interactions between the dansyl units in the range of temperatures studied in this work. Lowering the temperature from 293 K to 213 K resulted in a lengthening the fluorescence decay times independent of the dendrimer generation (Table 1). Given the negligible temperature-effect on the shortest distance (∼2.9 Å) between dansyls and the G4 amine groups, the increase in lifetime is most probably due to slowed down solvent relaxation around the dansyl chromophores. As dansyl is reported to be insensitive to oxygen quenching,42 it is not expected to be behind the observed lifetime changes.Fluorescence anisotropy decays were best fitted by double exponentials. All anisotropy decays were slowed down as the temperature of the dendrimer solutions was reduced, indicating the temperature dependence of the internal and rotational dynamics of the dendrimers. In our previous study it was found that an almost non-existent spectral overlap of the dansyl absorption and fluorescence spectra prevents effective energy migration among dansyls and therefore the most likely reason for fluorescence anisotropy decay is the motions of the dansyl chromophores.10 The anisotropy decay of the relatively freely moving outer-sphere dansyls is dominated by fast local motions, whereas the back-folded dansyls rotate at slower pace.10 Time resolution of our TCSPC setup allows the resolution of these decays down to about 100 ps, which leaves the study of the fast motions out of reach. However, these motions, occurring in the time domain from 50 ps to 100 ps, were studied in our previous work where streak camera measurements were deployed.10 The faster anisotropy decays resolved in the present study are then likely to be convolutions of the dynamics of the dendrimer branches and the local motions of the dansyls. These motions slow down as temperature is lowered (Table 1). The slowing down is strongest for the G4 generation, indicating a freezing of these motions in this large dendrimer. The longer anisotropy decays are related to the overall rotation of the dendrimers. The lower the temperature of the dendrimer solution, the higher is the relative amplitude of the longer anisotropy component. Rotation of the whole dendrimer becomes the prevailing mechanism of the anisotropy decay at lower temperatures due to higher rigidity of the structure.
Rotation correlation times and volumes of the dendrimers
The lengthening of the rotational correlation times was observed for all generations of dendrimers. For G1 it went from 0.3 ns at 293 K to 1.58 ns at 213 K. The analogous values for higher generations were from 1.17 ns to 4.26 ns for G2, from 1.48 ns to 9.24 ns for G3 and from 2.61 ns to 19.1 ns for G4. The relative lengthening of the rotational correlation times for the G3 and G4 generations is higher than that for the G1 and G2 generations.With values ranging from 6.86 ns (213 K) to 1.55 ns (273 K), and despite the underestimated values due to partial inaccuracies of the adopted force-field,31 the simulations qualitatively reproduce the experimental lengthening at low temperatures (Table 4). As noted above, the calculated values can be rather accurately fitted to an exponential decay ln(τrot) = ln(τ0) − βT with β = 0.024 K−1. Notably, fitting of the experimental G4-dataset to the same function, yields β = 0.025 K−1. Thus, the calculated temperature-driven lengthening rate is in good agreement with the experiments, indirectly validating the choice of both the model and time-scale adopted in the simulations.
Besides the reduction of thermal energy, there are three main reasons for the slowing down of the overall rotation 1) higher viscosity of the solvent, 2) increased G4-solvent interactions (Fig. 7), and 3) increased statistical volume of the dendrimer. The latter effect may be separated by using the Stokes–Einstein–Debye (SED) equation to estimate the volumes of the dendrimers at various temperatures
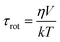 | (9) |
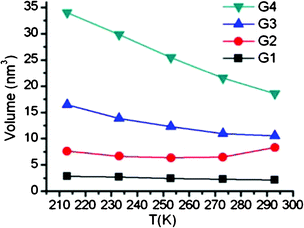 | ||
| Fig. 8 Temperature dependence of the G1–G4 dendrimer volumes calculated from the long anisotropy decay components by using hydrodynamic Stokes–Einstein–Debye equation. | ||
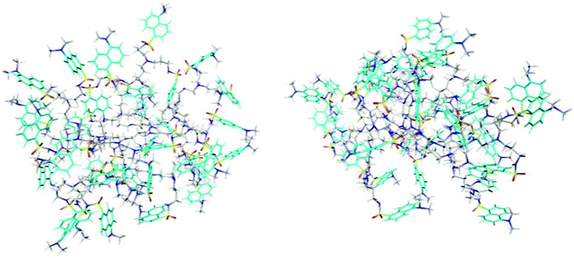 | ||
| Fig. 9 Two snapshots of the G4 dendrimer from MD simulations at 213 K (left) and 273 K (right). To better locate the dansyl groups, the naphthalene carbons are coloured in cyan. | ||
Shape of the dendrimer
An important issue to consider is the shape of the dendrimer. The SED equation is valid for spherical molecules. This assumption is not, however, entirely satisfactory with the dendrimers. Though theoretical calculations have predicted dendrimers to take more or less spherical shape as the generation grow,14,15,43 their exact shape is anything but straightforward to define. We used MD simulations to assess the shape of the G4 dendrimer. The ratios of the moments of inertia of G4 (Table 3) suggests that the shape of this dendrimer is slightly oblate, but still reasonably close to spherical, which is consistent with several studies.14,44 By calculating the asphericity ratio, also called relative shape anisotropy,14,15,45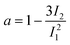 | (10) |
| I1 = Ix + Iy + Iz and I2 = IxIy + IxIz + IyIz | (11) |
Fluorescence of POPAM–eosin complexes
The dansyl fluorescence of the dendrimer–eosin complexes is efficiently quenched since EET from dansyls to eosins occurs in less than 10 ps.10,23 This value can be compared to the fluorescence lifetime of the free dendrimers of about 15 ns. The magic angle fluorescence lifetimes of the complex solutions recorded in the dansyl emission region are almost the same as the lifetimes of pristine dendrimers (cf.Tables 1 and 2). Hence it may be concluded that fluorescence from the complex solutions in the dansyl emission region arises entirely from the uncomplexed dendrimers. In G2 complexes, fluorescence is quenched to a lesser extent than in G4 complexes. Since the ratio of eosins to dansyls in all sample solutions was one to eight then the probability for exciting a G2 dendrimer without guest is much higher than the probability of exciting free G4 dendrimer.The generation dependent reduction of eosin fluorescence lifetimes (Table 2) in complexes, when compared to the eosin lifetime in solution of 3.6 ns,46 points to interaction between eosin(s) and the dendrimer. Partly non-radiative decay may take place from coupling of the eosin excited state to the dendrimer low frequency modes. The higher dendrimer generations G3 and G4 may have several eosins attached to the same dendrimer22 and the probability of EET between the eosins increases, which is seen in additional shortening of the fluorescence lifetimes in the G3 and G4 complexes. The G2 complex may host only one eosin and EET between the eosins does not occur (Table 2). The excitation of eosins directly at 480 nm or excitation of the dansyl moiety at 385 nm gave almost identical sub 5 ns fluorescence kinetics, which suggests that EET from dansyls to eosin(s) takes place much faster than the time resolution of the present experiments, as verified in our previous work.10,23 At 375 nm excitations, 5 per cent amplitude of 15 ns fluorescence from dansyls were resolved in the eosin spectral detection window, which is due to partial overlap of the dansyl and eosin emission spectra in this wavelength region.
Anisotropy decay of the complexes
After excitation of dansyls, the anisotropy signal could not be recorded in the eosin fluorescence region since it decays faster than the time resolution of our experiment due to EET from dansyls to eosin.10,23 After direct excitation of eosins with 485 nm pulses, the initial anisotropy of eosin fluorescence of the G2 complex was about 0.4 and the anisotropy decayed in about one nanosecond, a time scale similar to the anisotropy decay of the free G2 dendrimer. This finding suggests that eosin is rotating together with the dendrimer, since the anisotropy decay of the eosin in solution is clearly faster. For the G3 and G4 complexes the initial anisotropy was much lower. In G3 and G4 complexes, the low initial anisotropy values point towards a fast non-radiative decay process that is faster than the time resolution of the TCSPC experiment. Since both these complexes may have more than one encapsulated eosin per dendrimer the process is likely to be related to EET between eosins in the complexes.Conclusions
We have studied the dynamics of the dansylated POPAM dendrimers at several temperatures by time-resolved fluorescence measurements and molecular dynamics simulations. The results demonstrate that the volume and shape of the generation three and four dansylated POPAM dendrimers in solution are sensitive to temperature. At low temperatures the structure of the dendrimer is more rigid and peripheral dansyl groups do not fold back towards the centre of the molecule as readily as they do at room temperature. Thus, the overall rotation of the dendrimer molecule becomes the predominant anisotropy decay mechanism. The volume of first and second generation dendrimers is practically insensitive to temperature variation since the shorter branches of smaller dendrimers do not allow large back-folding. The dependence of the dendrimer volume on the temperature is found to originate from the balance between intra-dendrimer and short-range solvent–dendrimer interactions, with marginal contributions from the entropic gain due to the solvent molecules leaving the dendrimer. The phenomenon depends on the dendrimer generation, its functional groups and the type of the solvent. These findings open the way to the design of dendrimers with temperature-dependent volume, accessible area, and host–guest chemistry.We observed that the fluorescence lifetimes of the POPAM dendrimers were practically independent of the dendrimer generation in both the pure and complexed dendrimers. This is an expected result since excitation energy transfer from dansyls to eosin in the complexes is much faster than the radiative lifetime of the dansyls. For this reason no size and shape information was obtained from the POPAM–eosin complexes. Both the fluorescence lifetime and the initial anisotropy of the guest molecule eosin depended on the generation of the host dendrimer suggesting two non-radiative relaxation channels, excited eosin–dendrimer interaction and eosin–eosin EET in the higher generation POPAM's G3 and G4.
Acknowledgements
The research group of Professor Fritz Vögtle at University of Bonn is gratefully acknowledged for the synthesis of the dansylated POPAM dendrimers. GT is supported by EPSRC-UK (EP/I004483/1). JKT acknowledges financial support under from FinNano research program supported by the Academy of Finland (Contract No 118040) and the research grant under Academy of Finland (Contract No 123801).References
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117 CrossRef CAS.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003 CrossRef CAS.
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857 CrossRef CAS.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS.
- W. Jang, K. M. K. Selim, C. Lee and I. Kang, Prog. Polym. Sci., 2009, 34, 1 CrossRef CAS.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665 CrossRef CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517 CrossRef CAS.
- J. Aumanen, T. Kesti, V. Sundström, G. Teobaldi, F. Zerbetto, N. Werner, G. Richardt, J. van Heyst, F. Vögtle and J. Korppi-Tommola, J. Phys. Chem. B, 2010, 114, 1548 CrossRef CAS.
- E. K. L. Yeow, K. P. Ghiggino, J. N. H. Reek, M. J. Crossley, A. W. Bosman, A. P. H. J. Schenning and E. W. Meijer, J. Phys. Chem. B, 2000, 104, 2596 CrossRef CAS.
- V. Vicinelli, G. Bergamini, P. Ceroni, V. Balzani, F. Vögtle and O. Lukin, J. Phys. Chem. B, 2007, 111, 6620 CrossRef CAS.
- M. Maus, R. De, M. Lor, T. Weil, S. Mitra, U. Wiesler, A. Herrmann, J. Hofkens, T. Vosch, K. Müllen and F. C. De Schryver, J. Am. Chem. Soc., 2001, 123, 7668 CrossRef CAS.
- P. K. Maiti, T. Cagin, G. Wang and W. A. Goddard, Macromolecules, 2004, 37, 6236 CrossRef CAS.
- J. S. Klos and J.-U. Sommer, Macromolecules, 2009, 42, 4878 CrossRef CAS.
- D. A. Markelov, S. V. Lyulin, Y. Y. Gotlib, A. V. Lyulin, V. V. Matveev, E. Lähderanta and A. A. Darinskii, J. Chem. Phys., 2009, 130, 044907 CrossRef.
- S. De Backer, Y. Prinzie, W. Verheijen, M. Smet, K. Desmedt, W. Dehaen and F. C. De Schryver, J. Phys. Chem. A, 1998, 102, 5451 CrossRef CAS.
- P. K. Maiti, T. Cagin, S. Lin and W. A. Goddard, Macromolecules, 2005, 38, 979 CrossRef CAS.
- I. Tanis and K. Karatasos, Phys. Chem. Chem. Phys., 2009, 11, 10017 RSC.
- D. A. Markelov, V. V. Matveev, P. Ingman, M. N. Nikolaeva, E. Lähderanta, V. A. Shevelev and N. I. Boiko, J. Phys. Chem. B, 2010, 114, 4159 CrossRef CAS.
- F. Vögtle, S. Gestermann, C. Kauffmann, P. Ceroni, V. Vicinelli, L. De Cola and V. Balzani, J. Am. Chem. Soc., 1999, 121, 12161 CrossRef.
- V. Balzani, P. Ceroni, S. Gestermann, M. Gorka, C. Kauffmann and F. Vögtle, Tetrahedron, 2002, 58, 629 CrossRef CAS.
- J. Aumanen, V. Lehtovuori, N. Werner, G. Richardt, J. van Heyst, F. Vögtle and J. Korppi-Tommola, Chem. Phys. Lett., 2006, 433, 75 CrossRef CAS.
- A. Archut, S. Gestermann, R. Hesse, C. Kauffmann and F. Vögtle, Synlett, 1998, 546 Search PubMed.
- J. W. Powder, Tinker, Software Tools for Molecular Dynamics; 3.8Washington University School of Medicine: Saint Louis, MO, 2000 Search PubMed.
- G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2003, 125, 7388 CrossRef CAS.
- G. Teobaldi and F. Zerbetto, J. Lumin., 2005, 111, 335 CrossRef CAS.
- G. Teobaldi, M. Melle-Franco and F. Zerbetto, J. Chem. Theory Comput., 2005, 1, 194 CrossRef CAS.
- J. H. Lii and N. L. Allinger, J. Comput. Chem., 1998, 19, 1001 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Di Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- M. Dracinsky and P. Bour, J. Chem. Theory Comput., 2010, 6, 288 CrossRef CAS.
- C. E. Kundrot, J. W. Ponder and F. M. Richards, J. Comput. Chem., 1991, 12, 402 CrossRef CAS.
- M. L. Connolly, J. Appl. Crystallogr., 1983, 16, 548 CrossRef CAS.
- M. L. Connolly, J. Am. Chem. Soc., 1985, 107, 1118 CrossRef CAS.
- H. Goldstein, Classical Mechanics 2nd Ed., Addison-Wesley, Reading MA, 1980 Search PubMed.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes 3rd Ed., Cambridge University Press, Cambridge, 2007 Search PubMed.
- P. Atkins and J. De Paula, Atkins' Physical Chemistry 9th Ed., Oxford University Press, Oxford UK, 2010 Search PubMed.
- A. S. Rodgers, J. Chao, R. C. Wilhoit and B. J. Zwolinski, J. Phys. Chem. Ref. Data, 1974, 3, 117 CAS.
- N. L. Lange, Lange's Handbook of Chemistry 16th Ed., McGraw-Hill, New York, 2005 Search PubMed.
- Y. Sheng, S. Jiang and H. Tsao, Macromolecules, 2002, 35, 7865 CrossRef CAS.
- F. Vögtle, S. Gestermann, C. Kauffmann, P. Ceroni, V. Vicinelli and V. Balzani, J. Am. Chem. Soc., 2000, 122, 10398 CrossRef.
- C. M. Himel and R. T. Mayer, Anal. Chem., 1970, 42, 130 CrossRef CAS.
- M. L. Mansfield and L. I. Klushin, Macromolecules, 1993, 26, 4262 CrossRef CAS.
- H. Lee, J. R. Baker and R. G. Larson, J. Phys. Chem. B, 2006, 110, 4014 CrossRef CAS.
- D. N. Theodorou and U. W. Suter, Macromolecules, 1985, 18, 1206 CrossRef CAS.
- G. R. Fleming, A. W. E. Knight, J. M. Morris, R. J. S. Morrison and G. W. Robinson, J. Am. Chem. Soc., 1977, 99, 4306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00625h |
| This journal is © The Royal Society of Chemistry 2011 |