A facile and high-yield approach to synthesize one-dimensional CeO2 nanotubes with well-shaped hollow interior as a photocatalyst for degradation of toxic pollutants
Zi-Rong
Tang
*a,
Yanhui
Zhang
ab and
Yi-Jun
Xu
ab
aCollege of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, 350108, P.R. China. E-mail: zrtang@fzu.edu.cn; Tel: +86 591 22866126
bState Key Laboratory Breeding Base of Photocatalysis & College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, 350002, P.R. China
First published on 26th October 2011
Abstract
A facile, template-free and high-yield synthesis of single-crystalline cerium dioxide nanotubes (CeO2-NT) has been reported via a “casually-modified” approach based on the hydrothermal treatment of Ce(OH)CO3 precursors with alkali solution in an aqueous phase. This simple modification in synthesis procedures not only improves the yield of CeO2-NT remarkably, but also gives rise to the formation of CeO2-NT featuring excellent nanotubular open-ended structure with a well-shaped hollow interior. The collection techniques of BET, UV/visible diffuse reflectance spectra (DRS), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analysis have been employed to characterize the morphology and optical properties of the as-prepared CeO2-NT. Significantly, we demonstrate that CeO2-NT exhibits a markedly enhanced photocatalytic activity and stability as compared with its counterpart of CeO2 nanoparticles and commercial TiO2 (P25) toward the degradation of aromatic benzene, a well-known toxic pollutant that commonly occurs in urban ambient air and is of significant concern regarding environmental health because of its toxic, mutagenic, or carcinogenic properties. This represents a first example to demonstrate the advantage of CeO2 nanotubes as photocatalyst as compared to its counterpart of CeO2 nanoparticles, clearly suggesting the morphology/shape-dependent photocatalytic behaviour of CeO2 materials. Therefore, our current work not only offers a simple approach for fabrication of open-ended CeO2-NT with well-shaped hollow interior, but also demonstrates the promising potential of the applications of CeO2-NT, CeO2-NT-based and other metal oxide nanotube-based materials in the area of photocatalysis, which will inevitably enrich the intriguing chemistry of morphology/shape-dependent heterogeneous photocatalysis and thermal catalysis.
Introduction
One-dimensional organic and inorganic nanostructures including nanotubes, nanorods and nanowires have been attracting tremendous interest owing to their unique structural and electronic properties. To date, they have found broad applications in numerous areas, for example, light waveguides, solar energy conversion, catalysis, lithium battery, resonant Raman spectra, biomedical implants and gas sensing.1–9 In particular, one-dimensional nanotubes have well-defined hollow interiors that allow for the introduction of other matter including metal oxides, metal halides and organic molecules into the cavities, thus leading to nanotube-based composite materials with intriguing properties different from the bulk.10–13 For example, Bao and co-workers reported that a synergetic confinement of Rh metal particles inside the channel of carbon nanotubes results in a striking enhancement of catalytic activity for the conversion of CO and H2 to ethanol.14,15 Noteworthily, metal oxide nanotubes not only feature the special one-dimensional tubular nanostructure similar to carbon nanotubes, but also have the intrinsic chemical properties of metal oxides that are quite different from carbon nanotubes, which therefore attracts a great deal of scientific interest in controlled synthesis of CNT-like metal oxide nanotubes and exploration of their potential applications in many technological fields, for example, catalysis, electronic devices and gene delivery vehicles.16–20CeO2 is an important rare earth material because it is widely used for preparing catalysts, fuel cells, solar cells, UV blockers, polishing materials and hydrogen storage materials.21–29 It should be particularly noted that recent advances in the synthesis of CeO2 nanostructured materials with different morphologies, including nanospheres, nanorods, nanowires, nanocubes, nanopolyhedra, and nanotubes, offer new opportunities of enabling CeO2 and CeO2-based materials with desired structural and functional properties.30–36 For example, it has been shown that CeO2 nanotubes are very active for CO oxidation, and at 250 °C, the conversion rates of CO over CeO2 nanotubes are 3 times higher than that of the bulk counterpart, commercial ceria powder.37 So far, different methods have been reported to synthesize CeO2 nanotubes. Han et al. reported the production of CeO2 nanotubesvia a two-step procedure: precipitation at 100 °C and aging at 0 °C for 45 days.38 Tang et al. reported the synthesis of CeO2 nanotubes by annealing of Ce(OH)3 nanotubes prepared by an alkali thermal-treatment process under oxygen-free conditions.39 Zhou and co-workers reported a facile synthesis of CeO2 nanotubes with large cavities by etchingCe(OH)3 nanotubes/nanorods with H2O2.40 In addition to these template-free methods, the synthesis of CeO2 nanotubes using carbon nanotubes and porous alumina membrane as hard templates has also been reported.41,42 Most recently, Chen and co-workers reported the synthesis of three different types of CeO2 nanotubes by a hydrothermal treatment of Ce(OH)CO3 nanorods as precursor.37,43 Although significant progress has been achieved in the synthesis of CeO2 nanotubes based on a literature survey as mentioned above, there is still a lack of simple and effective methods for high-yield synthesis of open-ended CeO2 nanotubes with a well-shaped hollow interior that constitutes an important starting platform for anchoring other matter,10–15 such as noble metal particles and organic molecules, into the inside channel so as to investigate the synergetic confinement effect of CeO2 nanotubes. Furthermore, to the best of our knowledge, there is also no report on utilizing CeO2 nanotubes as photocatalysts for the degradation of environmental organic pollutants.
Herein, we report a facile template-free and high-yield synthesis of single-crystalline fluorite-structured cerium dioxide nanotubes (CeO2-NT) by a “casually-modified” approach based on the hydrothermal treatment of Ce(OH)CO3 precursors with alkali solution in an aqueous phase. This simple modification in synthesis procedure leads to the finding that the as-obtained CeO2-NT features an excellent nanotubular open-ended structure with a well-shaped hollow interior. Furthermore, the yield is remarkably enhanced. Significantly, we demonstrate for the first time that CeO2-NT exhibits an obviously enhanced photocatalytic activity as compared with its counterpart of CeO2 nanoparticles and commercial TiO2 (P25) toward the degradation of aromatic benzene, a well-known toxic pollutant that commonly occurs in urban ambient air and is of significant concern regarding environmental health because of its toxic, mutagenic, or carcinogenic properties.44–47 Thus, our present work not only offers a simple approach to synthesize open-ended CeO2 nanotubes with well-shaped hollow interior, but also demonstrates a promising potential of the applications of CeO2-NT, CeO2-NT-based and other metal oxide nanotube-based functional materials in the realm of photocatalysis, which consequently would enrich the chemistry of morphology/shape-dependent heterogeneous photocatalysis and thermal catalysis.
Experimental
Synthesis of CeO2 nanotubes
All reagents are of analytic grade and used without further purification. The synthesis of CeO2 nanotubes is via a “casually-modified” method based on the previous report regarding hydrothermal treatment of Ce(OH)CO3 precursors with alkali solution.37,43 The first step towards the synthesis of Ce(OH)CO3 precursors was based on the work reported by Chen et al.37 Typically, 1.7 g of Ce(NO3)3·6H2O and 3.6 g of urea were added to 80 mL of water under vigorous magnetic stirring for 30 min. Then, the clear suspension was transferred into a 100 mL round bottom flask for treatment in an oil bath at 80 °C for 24 h with stirring. After reaction, the suspension was cooled down to room temperature. The as-obtained powder samples were centrifuged and washed with distilled water, and dried completely in an oven by which the Ce(OH)CO3 precursors were obtained. The second step was the hydrothermal treatment of Ce(OH)CO3 precursors with NaOH solution in order to synthesize the CeO2 nanotubes. The key modification for this step was to improve the amount of Ce(OH)CO3precursors and the concentration of NaOH aqueous solution. This modification leads to the yield of the as-prepared CeO2 nanotubes being improved significantly; meanwhile, the as-synthesized CeO2 nanotubes feature a well-shaped hollow interior with open-ended structure. In a typical experiment, 0.25 g of the Ce(OH)CO3 precursors were redispersed into 40 mL of distilled water. Upon the addition of 4.8 g of NaOH, the mixture suspension was stirred for 30 min and then transferred into a 50 mL Teflon-lined stainless steel autoclave for a hydrothermal reaction at 120 °C for 24 h. After that, the products were cooled to room temperature, washed with distilled water and anhydrous ethanol, and then they were dried in air at 100 °C for 20 h. For comparison, the CeO2 nanoparticles of size 15–30 nm were supplied by Alfa Aesar.Characterization
The X-ray diffraction (XRD) analysis was performed on a Bruker D8 Advance X-ray diffractometer at 40 kV and 40 mA with Ni-filtered Cu-Kα radiation. The optical properties were analyzed by UV-vis diffuse reflectance spectroscopy (DRS) using a UV-vis spectrophotometer (Cary-500, Varian Co.), in which BaSO4 was used as the internal standard. The nitrogen adsorption–desorption isotherm was recorded using a Micromeritics-ASAP2020 instrument. The morphology of the samples was determined by field emission scanning electron microscopy (SEM) on a FEI Nova NANOSEM 230 spectrophotometer. Transmission electron microscopy (TEM) images were obtained using a JEOL model JEM 2010 EX instrument at the accelerating voltage of 200 kV. Electron spin resonance (ESR) spectra were measured using a Bruker A300 EPR electron paramagnetic resonance spectrometer equipped with a quanta-Ray Nd:YAG laser system as their radiation light source (λ = 254 nm). The settings were the center field at 3480.00 G, microwave frequency at 9.83 GHz, and power at 6.35 mW.Photocatalytic activity test
The gas-phase photocatalytic degradation of benzene was carried out in a tubular vessel microreactor operating in a continuous flow mode.45–47 All of the catalysts were sieved to obtain the particles of 0.21–0.25 mm size. Four 4 W UV lamps with wavelength centered at 254 nm (Philips, TUV 4W/G4 T5) were used as the light source because the as-prepared CeO2 nanotubes has the strongest light absorption around the region of 254 nm. A bubbler that contained benzene was immersed in an ice-water bath, and benzene (about 250 ppm) bubbled with oxygen from the bubbler was fed to 0.3 g catalyst at a total flow rate of 20 mL min−1. The reaction temperature was controlled at 30 ± 1 °C by an air-cooling system. Simultaneous determination of benzene and CO2 concentrations was performed with an online gas chromatograph (HP6890) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). Conversion of benzene was defined as follows: % conversion = [(Co − C)/Co] × 100 where Co is the initial concentration of benzene and C is the concentration of benzene after photocatalytic reaction.Results and discussion
Fig. 1 shows the X-ray powder diffraction (XRD) patterns of the as-prepared cerium dioxide nanotubes (CeO2-NT). It can be readily indexed to the pure cubic fluorite-structured CeO2 (JCPDS34-394), which is the same as that of bulk CeO2 nanoparticles. The Brunauer–Emmett–Teller (BET) adsorption–desorption isotherm of the as-synthesized CeO2-NT is shown in Fig. 2. The specific BET surface area and pore volume of CeO2-NT are 72 m2 g−1 and 0.14 cm3 g−1, respectively. The morphology and microscopic structure of CeO2-NT were disclosed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analysis, as displayed in Fig. 3. It is clear that the as-synthesized CeO2-NT sample has an inner diameter of around 10–25 nm along with a wall thickness of ca. 5–10 nm. Quite interesting is the observation from the TEM analysis that CeO2-NT consists of well-shaped and distinct hollow interior with open-ended structure. This structure character is evidently different from those reported in previous research works on the synthesis of CeO2 nanotubes.37–43 Thus, similar to the case of filling carbon nanotubes,10–15 this tubular CeO2-NT should be also suitable for introduction of noble metal particles, metal oxides, molten metal halides and organic molecules by nanocapillary wetting.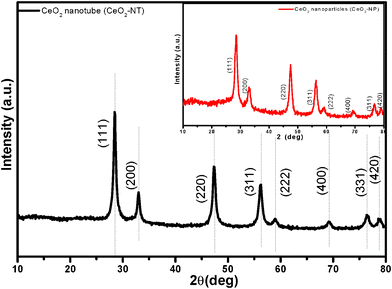 | ||
| Fig. 1 XRD pattern of the as-prepared CeO2 nanotubes (CeO2-NT); inset is the XRD pattern of CeO2 nanoparticles (CeO2-NP). | ||
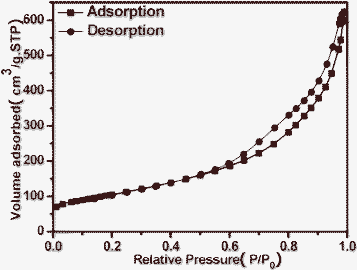 | ||
| Fig. 2 BET adsorption–desorption isotherm curve of the as-prepared CeO2 nanotubes (CeO2-NT). | ||
 | ||
| Fig. 3 Typical SEM (a), TEM (b, c, d) and HRTEM images (e, f) of the as-prepared CeO2 nanotubes (CeO2-NT). Inset is the selected area electron diffraction (SAED) pattern. | ||
The high-resolution transmission electron microscopy (HRTEM) analysis and selected area electron diffraction (SAED) pattern, as shown in Fig. 3f, reveal that the as-prepared CeO2-NT has a good single-crystalline structure, which is the same as that of L-type CeO2 nanotubes as reported by Chen et al.43 Noteworthily, our “casually-modified” approach not only gives rise to the formation of open-ended CeO2-NT with distinct well-shaped hollow interior, but also remarkably improves the synthesis yield of CeO2-NT that is beneficial for further exploration of its applications in catalysis. With a 50 mL autoclave, we can obtain about 0.21 g of CeO2-NT product per batch, which corresponds to a high yield of ca. 84% with reference to 0.25 g of Ce(OH)CO3 precursor as raw material.
The UV-vis diffuse reflectance spectra (DRS) are displayed in Fig. 4. It is clear that the sample of CeO2-NT possesses higher light absorption intensity than its counterpart of CeO2 nanoparticles (CeO2-NP) in both the UV and visible light region. Nevertheless, in the UV region, commercial P25 nanoparticles display higher light absorption capability than CeO2-NT and CeO2-NP. In addition, the light absorption edge of CeO2-NT displays a red shift as compared to the CeO2 nanoparticles. According to the Kubelka–Munk function transformation, the estimated band gap values of CeO2-NT, CeO2-NP and P25 are approximately 2.92 eV, 3.03 eV and 3.32 eV, respectively. These results suggest that the optical absorption property of CeO2 exhibits a morphology/shape-dependent behaviour and can be finely tailored by changing its microscopic morphology. The higher light absorption intensity of CeO2-NT than CeO2-NP also indicates that it may have enhanced photocatalytic performance for a specific target reaction. This inference is true as has been confirmed by the photocatalytic degradation of benzene in the gas phase as discussed below.
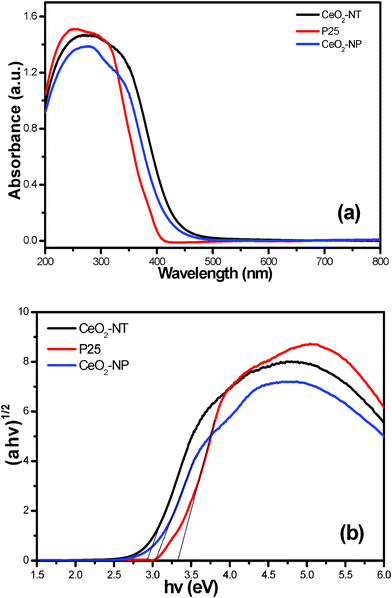 | ||
| Fig. 4 UV-vis diffuse reflectance spectra (DRS) of the as-prepared CeO2 nanotubes (CeO2-NT), CeO2 nanoparticles (CeO2-NP) and commercial P25 nanoparticles (a), and the plot of transformed Kubelka–Munk function versus the energy of light (b). | ||
Fig. 5 displays the time-online photocatalytic results for the gas-phase degradation of benzene over the samples of CeO2-NT, CeO2-NP and P25. As can be seen, during the reaction of 22 h, the sample of CeO2-NT exhibits the best photocatalytic performance toward the gas-phase degradation of benzene in view of the constant conversion ratio of benzene. Namely, no obvious deactivation trend is observed over the sample of CeO2-NT. In addition, after the reaction of 10 h, the produced amount of CO2 over CeO2-NT is nearly stable at 29 ppm. In contrast, the samples of both CeO2-NP and commercial P25-TiO2 nanoparticles display a quite unstable photocatalytic activity toward the gas-phase degradation of benzene. With regard to the sample of CeO2-NP, the conversion ratio of benzene at the initial stage is 2.2%; after reaction for 22 h, it is decreased to 1.4%. Furthermore, the produced amount of CO2 is remarkably decreased to only 8 ppm as compared to 19 ppm at the initial stage. In analogy, commercial P25 nanoparticles also exhibit the significant deactivation behaviour for the gas-phase degradation of benzene. The enhanced photocatalytic performance of CeO2-NT as compared to CeO2-NP seems consistent with its higher light absorption intensity as shown in UV-vis DRS spectra in Fig. 4. However, such a single correlation between light absorption intensity and photocatalytic activity is one-sided because other factors can also have an important and synergetic effect on photocatalytic performance, such as morphology, particle size and lifetime of photogenerated electron–hole pairs.45–47 Furthermore, it should be noted that P25 shows lower and more unstable activity than CeO2-NT, although it has higher light absorption intensity than CeO2-NT.
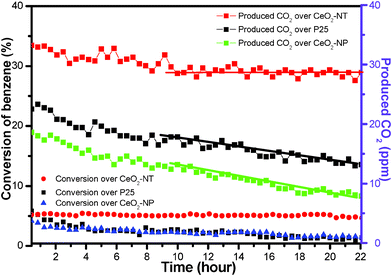 | ||
| Fig. 5 Time-online data for gas-phase photocatalytic degradation of benzene over the samples of commercial P25, CeO2 nanoparticles (CeO2-NP) and the as-prepared CeO2 nanotubes (CeO2-NT). | ||
To further understand why CeO2-NT shows more stable and higher photocatalytic activity than CeO2-NP and commercial P25, we have used electron spin resonance (ESR) spectra to observe the possible radical species formed over these samples under the irradiation of light. Information in this regard can help us learn the lifetime of electron–hole pairs and intensity of radical species, which are key photoactive species responsible for the degradation of benzene.45–47 The ESR data are shown in Fig. 6. It can be seen that the total intensity of hydroxyl radical (˙OH) species and superoxide radical (O2˙−) species as detected over the CeO2-NT sample is higher than that over the CeO2-NP and commercial P25 nanoparticles. In addition, it should be noted that, upon light irradiation, the O2˙− radical species are clearly observed over only the CeO2-NT sample but they are not observed over the samples of P25 and CeO2-NP. These suggest the prolonged lifetime of electron–hole pairs generated over the sample of CeO2-NT upon light irradiation. As a result, as reflected in Fig. 6, a greater amount of radical species with strong oxidation power, including ˙OH species and superoxide radical O2˙− is able to be photogenerated over the CeO2-NT than its counterpart CeO2-NP and commercial P25 nanoparticles. This in turn explains reasonably why the higher photocatalytic activity of CeO2 nanotubes than CeO2 and P25 nanoparticles has been observed toward the gas-degradation of benzene, as schematically shown in Fig. 7.
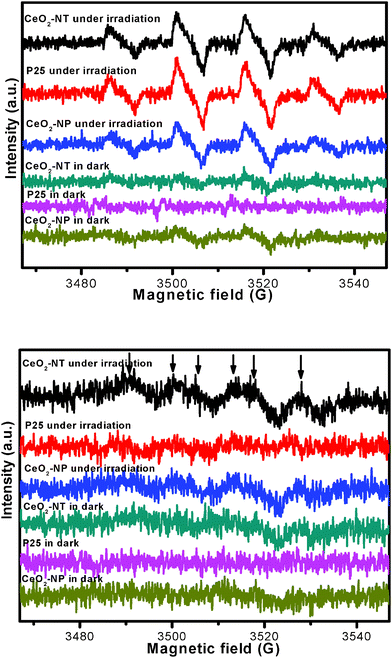 | ||
| Fig. 6 ESR spectra of radical adducts trapped by DMPO; (top) DMPO–˙OH radical species detected for the samples dispersion in water; (bottom) DMPO–O2˙− radical species detected for the sample dispersions in methanol. | ||
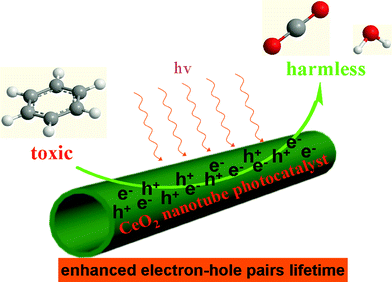 | ||
| Fig. 7 Proposed illustration showing the CeO2 nanotube as photocatalyst with enhanced electron–hole pair lifetime for degradation of the volatile organic pollutant benzene in the gas phase. | ||
In addition, the nanotubular morphology of CeO2-NT also imparts intrinsic advantages as a photocatalyst as compared with its counterpart of CeO2 nanoparticles, because it is well documented that: (1) the one-dimensional nanotube geometry facilitates the fast and long-distance electron transport; (2) the nanotubular morphology is beneficial for the enhancement of light absorption and scattering owing to the high length-to-diameter ratio.47–55
Our research work presents a first example that the photocatalytic activity of CeO2 can be tailored by changing its microscopic morphology. That is, one-dimensional nanotubular CeO2 possesses higher and more stable photocatalytic performance that bulk CeO2 nanoparticles toward the gas-phase degradation of the organic pollutant benzene in air. Such morphology/shape-dependent catalytic properties have been also observed previously in thermal heterogeneous catalysis. For example, the activity of CeO2 nanotubes has been shown to be three times higher than that of their bulk counterpart for CO oxidation.37 It has been demonstrated that the CeO2 nanorods are more reactive for CO oxidation than irregular nanoparticles of CeO2.56 In addition, nanopolyhedral CeO2 exhibit higher catalytic activity for CO oxidation than mesoporous CeO2 owing to the higher particle dispersity of the former than that of the latter.57 Therefore, it is reasonable to believe that, similar to morphology-dependent catalytic activity of CeO2 in thermal heterogeneous catalysis, there would be a wide scope to further tune the photocatalytic activity of semiconductor CeO2 through adjusting the morphology of CeO2 material by a variety of morphology-controlled synthesis approaches.
Conclusions
In summary, we have reported a facile, template-free and high-yield synthesis of single-crystalline cerium dioxide nanotubes (CeO2-NT) by a “casually-modified” approach based on the hydrothermal treatment of Ce(OH)CO3 precursors with alkali solution in an aqueous phase. This simple modification in synthesis procedure not only improves the yield of CeO2-NT remarkably, but also gives rise to the formation of CeO2-NT featuring excellent nanotubular open-ended structure with a well-shaped hollow interior, which could offer solid inroads into further anchoring other matter into the inside channel of CeO2 nanotubes. In addition, we have presented a first example that CeO2-NT exhibit a markedly enhanced photocatalytic activity as compared with its counterpart CeO2 nanoparticles and commercial TiO2 (P25) toward the degradation of aromatic benzene, a well-known toxic pollutant that commonly occurs in urban ambient air. This clearly illustrates the morphology/shape-dependent photocatalytic behaviour of CeO2 materials. Therefore, our work demonstrates the promising potential of the applications of CeO2-NT, CeO2-NT-based and other metal oxide nanotube-based materials in photocatalysis, which will therefore enrich the chemistry of morphology/shape-dependent functional properties related to CeO2 materials as heterogeneous photocatalysts. Finally, it is hoped that the current work will stimulate further interest in this intriguing topic.Acknowledgements
The support by the National Natural Science Foundation of China (20903022, 20903023), the Award Program for Minjiang Scholar Professorship, Program for Changjiang Scholars and Innovative Research Team in Universities (PCSIRT0818), National Basic Research Program of China (973 Program: 2007CB613306), the Science and Technology Development of Foundation of Fuzhou University (2009-XQ-10), the Open Fund of Photocatalysis Institute of Fuzhou University (0380038004), and Program for Returned High-Level Overseas Chinese Scholars of Fujian province is gratefully acknowledged.References
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS.
- Z. L. Wang, ACS Nano, 2008, 2, 1987–1992 CrossRef CAS.
- B. Tian, X. Zheng, T. J. Kempa, Y. Fang, N. Yu, G. Yu, J. Huang and C. M. Lieber, Nature, 2007, 449, 885–889 CrossRef CAS.
- A. I. Hochbaum, R. Chen, R. D. Delgado, W. Liang, E. C. Garnett, M. Najarian, A. Majumdar and P. Yang, Nature, 2008, 451, 163–167 CrossRef CAS.
- G. K. Mor, O. K. Varghese, M. Paulose, K. Shankar and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 2011–2075 CrossRef CAS.
- H. Zhou and S. S. Wong, ACS Nano, 2008, 2, 944–958 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- C. N. R. Rao, F. L. Deepak, G. Gundiah and A. Govindaraj, Prog. Solid State Chem., 2003, 31, 5–147 CrossRef CAS.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS.
- A. N. Khlobystov, D. A. Britz and G. A. D. Briggs, Acc. Chem. Res., 2005, 38, 901–909 CrossRef CAS.
- P. M. Ajayan and S. Iijima, Nature, 1993, 361, 333–334 CrossRef CAS.
- S. C. Tsang, Y. K. Chen, P. J. F. Harris and M. L. H. Green, Nature, 1994, 372, 159–162 CrossRef CAS.
- D. Ugarte, A. Chatelain and W. A. de Heer, Science, 1996, 274, 1897–1899 CrossRef CAS.
- X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo and X. Bao, Nat. Mater., 2007, 6, 507–511 CrossRef CAS.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS.
- R. Tenne, Nat. Nanotechnol., 2006, 1, 103–111 CrossRef CAS.
- Y. J. Xiong, B. T. Mayers and Y. N. Xia, Chem. Commun., 2005, 5013–5022 RSC.
- R. Tenne, Angew. Chem., Int. Ed., 2003, 42, 5124–5132 CrossRef CAS.
- C. N. R. Rao and A. Govindaraj, Adv. Mater., 2009, 21, 4208–4233 CrossRef CAS.
- X. Wang and Y. D. Li, Inorg. Chem., 2006, 45, 7522–7534 CrossRef CAS.
- A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 38, 439 CrossRef CAS.
- B. M. Reddy, P. Bharali and P. Saikia, J. Phys. Chem. C, 2007, 111, 1878–1881 CrossRef CAS.
- S. D. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS.
- X. J. Yu, P. B. Xie and Q. D. Su, Phys. Chem. Chem. Phys., 2001, 3, 5266–5269 RSC.
- D. G. Shchukin and R. A. Carus, Chem. Mater., 2004, 16, 2287–2292 CrossRef CAS.
- K. Sohlberg, S. T. Pantelides and S. F. Pennycook, J. Am. Chem. Soc., 2001, 123, 6609–6611 CrossRef CAS.
- G. Jacobs, L. Williams, U. Graham, D. Sparks and B. H. Davis, J. Phys. Chem. B, 2003, 107, 10398–10404 CrossRef CAS.
- A. Varez, E. Garcia-Gonzalez and J. Sanz, J. Mater. Chem., 2006, 16, 4249–4256 RSC.
- R. Di Monte and J. Kaspar, J. Mater. Chem., 2005, 15, 633–648 RSC.
- P. F. Ji, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2008, 112, 17809–17813 CrossRef CAS.
- L. Liao, H. X. Mai, Q. Yuan, H. B. Lu, J. C. Li, C. Liu and C. H. Yan, J. Phys. Chem. C, 2008, 112, 9061–9065 CrossRef CAS.
- N. Du, H. Zhang, B. D. Chen, X. Y. Ma and D. R. Yang, J. Phys. Chem. C, 2007, 111, 12677–12680 CrossRef CAS.
- W. Q. Han, L. J. Wu and Y. M. Zhu, J. Am. Chem. Soc., 2005, 127, 12814–12815 CrossRef CAS.
- S. W. Yang and L. Gao, J. Am. Chem. Soc., 2006, 128, 9330–9331 CrossRef CAS.
- H. P. Zhou, Y. W. Zhang, H. X. Mai, X. Sun, Q. Liu, W. G. Song and C. H. Yan, Chem.–Eur. J., 2008, 14, 3380–3390 CrossRef CAS.
- H. Li, G. Lu, Q. Dai, Y. Wang, Y. Guo and Y. Guo, ACS Appl. Mater. Interfaces, 2010, 2, 838–846 CrossRef CAS.
- G. Chen, C. Xu, X. Song, W. Zhao, Y. Ding and S. Sun, Inorg. Chem., 2008, 47, 723–728 CrossRef CAS.
- W. Q. Han, L. J. Wu and Y. M. Zhu, J. Am. Chem. Soc., 2005, 127, 12814–12815 CrossRef CAS.
- C. Tang, Y. Bando, B. Liu and D. Golberg, Adv. Mater., 2005, 17, 3005–3009 CrossRef CAS.
- K. B. Zhou, Z. Q. Yang and S. Yang, Chem. Mater., 2007, 19, 1215–1217 CrossRef CAS.
- K. Yu, G. L. Ruan, Y. H. Ben and J. J. Zou, Mater. Sci. Eng., B, 2007, 139, 197–200 CrossRef CAS.
- D. Zhang, H. Fu, L. Shi, J. Fang and Q. Li, J. Solid State Chem., 2007, 180, 654–660 CrossRef CAS.
- G. Chen, S. Sun, X. Sun, W. Fan and T. You, Inorg. Chem., 2009, 48, 1334–1338 CrossRef CAS.
- Q. Lan, L. Zhang, G. Li, R. V. Vermeulen, R. S. Weinberg, M. Dosemeci, S. M. Rappaport, M. Shen, B. P. Alter, Y. Wu, W. Kopp, S. Waidyanatha, C. Rabkin, W. Guo, S. Chanock, R. B. Hayes, M. Linet, S. Kim, S. Yin, N. Rothman and M. T. Smith, Science, 2004, 306, 1774–1776 CrossRef CAS.
- Y. J. Xu, Y. Zhuang and X. Fu, J. Phys. Chem. C, 2010, 114, 2669–2676 CrossRef CAS.
- Y. Zhang, Z. R. Tang, X. Fu and Y. J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS.
- Z. R. Tang, F. Li, Y. Zhang, X. Fu and Y. J. Xu, J. Phys. Chem. C, 2011, 115, 7780–7886.
- K. Shankar, J. I. Basham, N. K. Allam, O. K. Varghese, G. K. Mor, X. Feng, M. Paulose, J. A. Seabold, K. S. Choi and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 6327–6359 CrossRef CAS.
- A. B. F. Martinson, J. E. McGarrah, M. O. K. Parpia and J. T. Hupp, Phys. Chem. Chem. Phys., 2006, 8, 4655–4659 RSC.
- Y. Ohsaki, N. Masaki, T. Kitamura, Y. Wada, T. Okamoto, T. Sekino, K. Niihara and S. Yanagida, Phys. Chem. Chem. Phys., 2005, 7, 4157–4163 RSC.
- T. Tachikawa, S. Tojo, M. Fujitsuka, T. Sekino and T. Majima, J. Phys. Chem. B, 2006, 110, 14055–14059 CrossRef CAS.
- J. R. Jennings, A. Ghicov, L. M. Peter, P. Schmuki and A. B. Walker, J. Am. Chem. Soc., 2008, 130, 13364–13372 CrossRef CAS.
- K. Zhu, T. B. Vinzant, N. R. Neale and A. J. Frank, Nano Lett., 2007, 7, 3739–3746 CrossRef CAS.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS.
- T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2009, 131, 8485–8495 CrossRef CAS.
- K. B. Zhou, X. Wang, X. M. Sun, Q. Peng and Y. D. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS.
- H. P. Zhou, Y. W. Zhang, R. Si, L. S. Zhang, W. G. Song and C. H. Yan, J. Phys. Chem. C, 2008, 112, 20366–20374 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |