Hierarchical structures formed from self-complementary sextuple hydrogen-bonding arrays†
Chih-Chia
Cheng
,
Ying-Chieh
Yen
and
Feng-Chih
Chang
*
Institute of Applied Chemistry, National Chiao-Tung University, 30050, Hsinchu, Taiwan. E-mail: changfc@mail.nctu.edu.tw; Fax: +886-3-5131512
First published on 29th September 2011
Abstract
A new self-complementary supramolecule based on U-DPy possesses an extremely high association constant, far exceeding those of existing systems (Kdimer > 107 M−1). In addition, its sextuple hydrogen bonding interaction and π-packing can be further promoted through annealing, resulting in a flat ribbon structure.
Supramolecular self-assembly has attracted great attention because of the novel structural organizations formed through highly complementary molecular recognition events.1 A typical example is the assembling and disassembling of structural motifs such as DNA and RNA—fundamental for translation and transcription in biology, offering unique opportunities for enhanced control of self-assembly.2 These noncovalent interaction containing materials exhibit unique physical properties, such as high specificity, controlled affinity, and reversible, selective, self-healing, and spontaneous self-assembly behavior.3 Recently, several supramolecular structures have been incorporated into polymers4 to form novel materials with features of conventional polymers while featuring reversibility in the bonding between monomer units.5 On the other hand, chemists have started to learn not only how to mimic Nature's use of noncovalent chemistry in polymer science but also how we might be able to harness these interesting biological molecules to construct complex nanostructures and materials in the foundation of supramolecular polymer science.
Noncovalent interactions such as hydrogen bonding, ionic interactions, metal coordination, electrostatic interactions, and π–π stacking have been used individually or in concert with one another to obtain well-controlled and reliable nanostructures.6Hydrogen bonding interactions are the most employed noncovalent interactions for functionalizing polymeric scaffolds5,7 because the strength of the hydrogen bonded complexes can be tuned easily by altering the donor (D) and acceptor (A) moieties. Typically, the hydrogen bonding strength in stabilized complementary complexes can be tuned by varying the number of hydrogen bonds from single,8a double,8b,c triple,8d quadruple8e–g to sextuple or even higher order hydrogen bonding motifs.8h–k By choosing an appropriate hydrogen bonding motif, the strength of the bonding with binding constant larger than 106 M−1 can be obtained, which is close to the strength of conventional covalent linkages.1,9 More recently, Meijer and co-workers10 demonstrated concentration dependent selectivity for the heterodimerization of 2,7-diamide-1,8-naphthyridine (DAN) derivatives8e and 2-ureido-6-pyrimidinone (UPy)8f,g Furthermore, employing bifunctional DAN and UPy motifs is advantageous because this AB-type monomer is able to form cycle structures at low concentrations, and leads to the formation of a supramolecular polymer by increasing the concentration.10c In addition, the relevant interaction relies on the Hamilton receptor/barbituric acid interaction, which is able to assemble via a strong six-point hydrogen bond association.8h,i Lehn11a and Binder11b used the hydrogen bonding motif in combination for the preparation of chain-extended supramolecular polymers and block copolymers. Nevertheless, controlling the secondary (and higher) structures of synthetic polymers remains a challenging task.12
Recently, we have reported the biocomplementary interactions between a nucleobase-like side-chain homopolymer and alkylated nucleobases mediated by thymine–adenine and uracil–adenine base pairs.13 In this study, new derivatives of N-(6-(3-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)propanamido)pyridin-2-yl)undec-10-enamide (U-DPy) dimerizing easily and strongly by means of a self-complementary DADADA array of sextuple hydrogen bonding interactions were developed. This new derivative possesses unique stable associating behavior and self-assembled structures.
Synthesis and self-complementary behavior of a new U-DPy . Herein we developed a new heterocyclic U-DPy derivative with alkyl groupsvia a three-step synthesis as shown in Scheme 1, which possesses self-complementary DADADA, spatial arrangement, and tunable solubility.
 | ||
| Scheme 1 Synthetic procedures used to obtain U-DPy and A-DPy. | ||
The monoamide 1 was obtained by monoamidation of 2,6-diaminopyridine with undecanoyl chloride. Acylation of the amino group of 1 yielded acrylated 3, and then the final product was obtained by Michael addition of the acrylated 3 with uracil as previously described.14 Most importantly, the observed 1H NMR spectrum of U-DPy in CDCl3 (see ESI,† Fig. S1) showed a clear downfield peak (12.67 ppm) of the N–H proton (of uracil units), which is consistent with the formation of the selective hydrogen bonded complex between uracil and diamidopyridine (DAP).15 In a control experiment, the 1H NMR spectrum of A-DPy showed that these peaks did not shift in CDCl3 as compared with U-DPy (Scheme 1 and Fig. S4). To investigate the stability of the hydrogen bonding interaction, we performed variable-temperature NMR experiments in toluene-d8 solutions (Fig. 1).
 | ||
| Fig. 1 1H NMR spectra at various temperatures for a 40 mM solution of U-DPy in toluene-d8. | ||
When the solution containing 40 mM of U-DPy was heated from 30 to 105 °C, the peak of the amide proton was slightly upfield shifted from 12.32 ppm at 30 °C to 12.16 ppm at 105 °C. This observation indicates that the hydrogen bonding interaction was not appreciably destroyed during heating and the supramolecular polymerization did not occur through the triple hydrogen bonding interactions (Scheme 2), which was rarely observed. In the previous study, the multiple hydrogen bonding interaction was influenced with increasing temperature, showing an obvious shift of amide proton signals (Fig. S7 and S8)16 In addition, the entropically favored unfolding structure possessing steric effect tends to hinder the formation of the intramolecular hydrogen bonds, the intermolecular sextuple hydrogen bonding arrays of U-DPy are depicted in Scheme 2.
 | ||
| Scheme 2 The proposed hydrogen bonding mechanism of U-DPy as formation of a dimer. | ||
In an attempt to discuss the formation of intermolecular hydrogen bonding interactions, the concentration-dependent selectivity was also examined. 1H NMR spectra did not show any apparent change in chemical shifts across a broad concentration range (100 mM to 0.07 mM) of U-DPy in dilute CDCl3 solution (Fig. S3). Since the chemical shifts did not change and no new peaks appeared, the dimerization constant (Kdimer) of U-DPy was estimated to be >107 M−1.8f,17 Therefore, we speculate that the sextuple hydrogen bonding interaction may possesses the largest dimerization constant to date for neutral, hydrogen-bonded species.
Molecular recognition in pyrene -based U-DPy self-complexes. In order to more accurately establish the dimerization constant (Kdimer) of U-DPy, a pyrene-labeled compound, U-DPy-Pyrene, was synthesized and studied by fluorescence spectroscopy (see ESI† for details). Pyrene-based fluorescence exhibits a large extinction coefficient, excellent quantum yield, and good stability in aqueous solution. Pyrene also forms an excited state dimer, excimer, with readily detected emission that is a red shift of ca. 100 nm from the monomer.18a Therefore, pyrene has been employed to determine the Kdimer value in solution by monitoring its excimer intensity with varied concentration.18b,c To evaluate the influence of solvents on hydrogen bonding interactions and determine the solvent dependence of intermolecular excimer formation, 10−5 M solutions of U-DPy-Pyrene were dissolved in several (hydrophobic and hydrophilic) solvents (Fig. 2a). The excimer emission intensity varied substantially with different solvents. Those hydrophobic solvents (toluene, chloroform and ethyl acetate) deliver most of the excimer emission, whereas hydrophilic solvents (THF, DMF and DMSO) weaken the excimer signal.19 To understand how these excimers were formed in hydrophobic solvents, the fluorescence was measured in a 10−5 M solution of U-DPy-Pyrene in the presence of a 30-fold excess of the U-DPy (non-emissive) in chloroform where the intermolecular excimer formation did not occur. The excimer can be extracted due to the formation of the hetero-dimer (U-DPy:U-DPy-Pyrene) (Fig. 2b). In order to determine the dimerization constant of the U-DPy-Pyrene, fluorescence spectra were measured with various concentrations (10−10 to 10−4 M) of U-DPy-Pyrene in chloroform as previously reported.18c Surprisingly, the decrease in the concentration of U-DPy-Pyrene led to an increase in corrected excimer intensity (I*) (Fig. 2c and S9), whereas pure pyrene exhibited the opposite trend. The observation indicates that the hydrogen bond strength of the U-DPy-Pyrene homo-dimer formation is stronger than current techniques based on fluorescence and the U-DPy-Pyrene form highly stable hydrogen-bonded complexes in chloroform. Although the exact Kdimer value has not yet been successfully obtained via the above mentioned methods, great efforts to measure the Kdimer of the dimeric species are in progress in our laboratory. In addition, the intriguing hydrogen bond feature led us to investigate the self-assembly of this supermolecule in the bulk state.
 | ||
| Fig. 2 Fluorescence experiments. (a) Excimer behavior in 10−5 M solutions of U-DPy-Pyrene homo-dimer. Emission spectra are normalized to the monomer peak at approximately 398 nm. (b) Constant (10−5 M) concentration of U-DPy-Pyrene in chloroform, mixed with increasing concentrations of non-emissive U-DPy. (c) Normalized fluorescence at differential concentrations of U-DPy-Pyrene in chloroform. | ||
Self-assembly of U-DPy in bulk state. Supramolecular self-assembly can be used as a bottom-up approach for the development of advanced structures.20Fig. 3a illustrates FTIR spectra of the N–H stretching region of the U-DPy. Vibration at 3278 cm−1 corresponds to the stretching of the medium hydrogen-bonded N–H groups, and the characteristic peaks at 3115 and 3205 cm−1 are frequencies generally observed for strongly hydrogen-bonded N–H.21a,b Notably, an absorption peak above 2500–2750 cm−1 occurs, indicative of N–H⋯N inter-association,21c which implies that uracil groups are highly complementary to the DAP group. Upon heating from 30 to 160 °C, the peaks at 3173 and 3205 cm−1 slightly shift to higher wavenumber and the amide N–H stretching vibration at 3278 cm−1 appears gradually. However, the free amide N–H group (3400–3600 cm−1) was not observed upon the heating process, suggesting that the U-DPy formed an extremely stable, self-complementary hydrogen-bonded complex in the bulk state. In the lower wavenumber region (1400–1750 cm−1) of the FT-IR spectra (Fig. 3b), three carbonyl stretching peaks were observed at 30 °C, peaks at 1680 and 1705 cm−1 correspond to the presence of the strongly associated N–H groups.21d As the temperature was increased from 80 to 100 °C , a new peak at 1667 cm−1 appeared with the absence of the peak at 1680 cm−1, suggestive of a change in the nature of the hydrogen bonding. Further investigation on this transition behavior was carried out using wide angle X-ray scattering (WAXS) measurements. The pristine U-DPy exhibited sharp crystal halos centered at q = 2.64 nm−1 as shown in the WAXS patterns in Fig. S10. Surprisingly, annealing U-DPy resulted in the appearance of a well-ordered regularization (Fig. 3c). The first intense reflection was observed at q = 2.15 nm−1 corresponding to a spacing of d = 2.95 nm, which is close to a basis of the molecular structure calculation (1.51 + 1.33 nm) (Scheme 2) and high order lamella microstructures were also observed (n = 1 up to 5). In addition, the WAXS pattern in Fig. 3c displays two reflection peaks at q = 7.35 (d = 0.85 nm) and q = 17.78 (d = 0.35 nm). The d spacing of 0.35 nm is consistent with the presence of π–π stacking between the dimeric layers. Generally, such interactions occur only in stabilized sheets.22a–c The d spacing of 0.85 nm suggests that there are stacks of the dimer that are held together through π–π interactions within these interaction domain.22b,c The interlayer distance in the heterocyclic core of q = 13.7–16.5 (d = 0.36–0.46 nm) is similar to that reported as π-stacking.22d–f As a control experiment, pristine and annealed A-DPy (Fig. S11) displayed peaks at q = 5.54 and 5.33 nm−1, respectively (d = 1.13 and 1.77 nm, respectively), indicative of unspecified structures at a relatively large size scale. The observed spacing indicated that the intermolecular A⋯DAP interactions did not occur and the self-assembly behavior was not observed. Based on these variable-temperature FTIR and WAXS analyses, the annealing-induced π–π interactions of U-DPy would form a new regular lamellar microstructure which differs from that of the pristine state as depicted in Fig. 3d.
 | ||
| Fig. 3 Variable-temperature FTIR spectra of the U-DPy presented in the ranges (a) 2300–3600 and (b) 1600–1750 cm−1. (c) WAXS datum for annealing U-DPy and (d) graphical representations of the lamellar structures of the self-complexes in bulk state. | ||
The self-assembly structures of annealed U-DPy in the bulk state were observed through scanning electron microscopy (SEM) and atomic force microscopy (AFM). Flat ribbon-like structures were observed from both analyses as the solution of U-DPy in chloroform (2.2 × 10−4 M) was dried and annealed on glass (Fig. 4). The dimensions of these flat ribbons were uniform and the thickness and width were 60.0 and 600.0 nm, respectively. In addition, the length of the ribbon length was of over 10 μm (Fig. 4b and 4d) while its width and thickness remained constant. The AFM images in Fig. 4d and 4e indicate that the microstructures are well arranged in lamellar-like ribbons organized through the side-by-side flat packing of the ribbons.23 Therefore, a self-organization mechanism was proposed from the annealed U-DPy as shown in Scheme 3, which was constructed on lamellae microstructures from the WAXS patterns. The close π-packing of these lamellae with d spacing of 2.95 nm forms flat ribbon structures, and then the ribbon-shaped aggregates are formed by single ribbons of 60 nm width placed side by side.
 | ||
| Fig. 4 SEM (a, b) and AFM (c–e)images showing the flat ribbon-like aggregates of annealed U-DPy, as indicated by the arrows in (b), to fit the thickness and width. AFM (d) is a magnified image of the squared region in (c), as indicated by the arrows, to fit the thickness. (e) 3D representation of the surface shown in (d). | ||
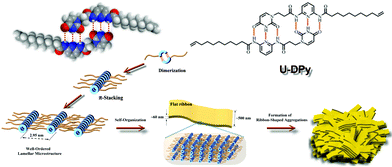 | ||
| Scheme 3 Suggested processes of the formation of the ribbon-shaped aggregates in the bulk state. | ||
In summary, a new self-complementary supramolecule based on U-DPy was successfully synthesized and assembled through intermolecular sextuple hydrogen-bonding arrays. The sextuple hydrogen-bonding interaction possesses the largest dimerization constant to date for neutral, hydrogen-bonded species (Kdimer > 107 M−1) and can be further promoted through annealing. In addition, annealingU-DPy constructs a flat ribbon structure from the sextuple hydrogen bonding interaction and π-packing. The study of incorporating the sextuple hydrogen bonded group into polymers on elucidating the factors affecting supramolecular polymer control and the development in materials with practical applications are continuing. Our further results will be reported in due course.
References
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS; (b) J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314 RSC.
- W. H. Binder and R. Zirbs, Adv. Polym. Sci., 2007, 207, 1 CrossRef CAS.
- C. R. South, C. Burd and M. Weck, Acc. Chem. Res., 2007, 40, 63 CrossRef CAS.
- R. Hoogenboom, D. Fourniera and U. S. Schubert, Chem. Commun., 2008, 155 RSC.
- A. J. Wilson, Soft Matter, 2007, 3, 409 RSC.
- S. Rieth, C. Baddeley and J. D. Badjic, Soft Matter, 2007, 3, 137 RSC.
- G. Armstrong and M. Buggy, J. Mater. Sci., 2005, 40, 547 CrossRef CAS.
- (a) E. Yoshida and S. Kunugi, Macromolecules, 2002, 35, 6665 CrossRef CAS; (b) L. L. De Lucca Freitas and R. Stadler, Macromolecules, 1987, 20, 2478 CrossRef; (c) C. Hilger, M. Draeger and R. Stadler, Macromolecules, 1992, 25, 2498 CrossRef CAS; (d) J. M. Pollino, K. P. Nair, L. P. Stubbs, J. Adams and M. Weck, Tetrahedron, 2004, 60, 7205 CrossRef CAS; (e) T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 11582 CrossRef CAS; (f) R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS; (g) J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef; (h) J. B. Bialecki, L.-H. Yuan and B. Gong, Tetrahedron, 2007, 63, 5460 CrossRef CAS; (i) V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. Eur. J., 2002, 8, 1227 CrossRef CAS; (j) B. A. Blight, C. A. Hunter, D. A. Leigh, H. McNab and P. I. T. Thomson, Nat. Chem., 2011, 3, 244 CrossRef CAS; (k) A. J. Wilson, Nat. Chem., 2011, 3, 193 CrossRef CAS.
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS; (b) A. Ciferri, Macromol. Rapid Commun., 2002, 23, 511 CrossRef CAS; (c) J.-M. Lehn, Polym. Int., 2002, 51, 825 CrossRef CAS; (d) S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC.
- (a) G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810 CrossRef CAS; (b) O. A. Scherman, G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11850 CrossRef CAS; (c) O. A. Scherman, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 2072 CrossRef CAS.
- (a) E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519 RSC; (b) W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. J. Kunz, Adv. Mater., 2005, 17, 2824 CrossRef CAS.
- M. Muthukumar, C. K. Ober and E. L. Thomas, Science, 1997, 277, 1225 CrossRef CAS.
- (a) C. C. Cheng, C. F. Huang, Y. C. Yen and F. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6416 CrossRef CAS; (b) C. C. Cheng, Y. C. Yen, Y. S. Ye and F. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2009, 43, 6388 CrossRef; (c) I. H. Lin, C. C. Cheng, Y. C. Yen and F. C. Chang, Macromolecules, 2010, 43, 1245 CrossRef CAS.
- A. S. Karikari, B. D. Mather and T. E. Long, Biomacromolecules, 2007, 8, 302 CrossRef CAS.
- P. Chattopadhyay and P. S. Pandey, Tetrahedron Lett., 2008, 49, 4640 CrossRef CAS.
- E. Kolomiets, E. Buhler, S. J. Candau and J.-M. Lehn, Macromolecules, 2006, 39, 1173 CrossRef CAS.
- (a) J. Rebek, Jr., B. Askew, P. Ballester, C. Buhr, S. Jones, D. Nemeth and K. Williams, J. Am. Chem. Soc., 1987, 109, 5033 CrossRef; (b) S.-K. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318 CrossRef CAS; (c) T. W. Bell and J. Liu, J. Am. Chem. Soc., 1988, 110, 3673 CrossRef CAS; (d) J. C. Adrian, Jr. and C. S. Wilcox, J. Am. Chem. Soc., 1989, 111, 8055 CrossRef; (e) T. J. Murray and S. C. Zimmerman, J. Am. Chem. Soc., 1992, 57, 4010 CrossRef; (f) M. R. Ghadiri, K. Kobayashi, J. R. Granja, R. K. Chadha and D. E. McRee, Angew. Chem., Int. Ed. Engl., 1995, 34, 93 CrossRef CAS; (g) P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 1998, 120, 9710 CrossRef CAS.
- (a) C. Garcia-Echeverria, J. Am. Chem. Soc., 1994, 116, 6031 CrossRef CAS; (b) P. S. Corbin, L. J. Lawless, Z. Li, Y. Ma, M. J. Witmer and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5099 CrossRef CAS; (c) S. H. M. Soentjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487 CrossRef CAS.
- (a) F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761 CrossRef CAS; (b) S. Zou, Z. Zhang, R. Foerch, W. Knoll, H. Schoenherr and G. J. Vancso, Langmuir, 2003, 19, 8618 CrossRef CAS.
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS; (b) T. Shimizu, M. Masuda and M. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS; (c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS; (d) R. F. Service, Science, 2005, 309, 95 CrossRef CAS.
- (a) J. Y. Lee, P. C. Painter and M. M. Coleman, Macromolecules, 1988, 21, 954 CrossRef CAS; (b) O. Uzun, A. Sanyal, H. Nakade, R. J. Thibault and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 14773 CrossRef CAS; (c) I. M. Vermeesch, G. Groeninckx and M. M. Coleman, Macromolecules, 1993, 26, 6643 CrossRef CAS; (d) Z. Sideratou, D. Tsiourvas, C. M. Paleos, E. Peppas, J. Anastassopoulou and T. Theophanides, J. Mol. Struct., 1999, 484, 91 CrossRef CAS.
- (a) V. R. Pedireddi, A. Ranganathan and K. N. Ganesh, Org. Lett., 2001, 3, 99 CrossRef CAS; (b) S. J. George and A. Ajayaghosh, Chem.–Eur. J., 2005, 11, 3217 CrossRef CAS; (c) S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202 CrossRef CAS; (d) H. J. Brouwer, V. V. Krasnikov, T. A. Pham, R. E. Gill, P. F. van Hutten and G. Hadziioannou, Chem. Phys., 1998, 227, 65 CrossRef CAS; (e) C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC; (f) R. Ziessel, G. Pickaert, F. Camerel, B. Donnio, D. Guillon, M. Cesario and T. Prange, J. Am. Chem. Soc., 2004, 126, 12403 CrossRef CAS.
- (a) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS; (b) E. Lee, J.-K. Kim and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 6375 CrossRef CAS; (c) M. Yang, W. Wang, I. Lieberwirth and G. Wegner, J. Am. Chem. Soc., 2009, 131, 6283 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00513h/ |
| This journal is © The Royal Society of Chemistry 2011 |