Magnetite/graphene nanosheet composites: interfacial interaction and its impact on the durable high-rate performance in lithium-ion batteries†
Jisheng
Zhou
a,
Huaihe
Song
*a,
Lulu
Ma
ab and
Xiaohong
Chen
a
aState Key Laboratory of Chemical Resource Engineering, Key Laboratory of Carbon Fiber and Functional Polymers, Ministry of Education, Beijing University of Chemical Technology, Beijing, P. R. China. E-mail: songhh@mail.buct.edu.cn; Fax: +86 10-64434916; Tel: +86 10-64434916
bDepartment of Mechanical Engineering and Materials Science, Rice University, Houston, TX 77005, USA
First published on 24th August 2011
Abstract
We explore in-depth the interfacial interaction between Fe3O4 nanoparticles and graphene nanosheets as well as its impact on the electrochemical performance of Fe3O4/graphene anode materials for lithium-ion batteries. Fe3O4/graphene hybrid materials are prepared by direct pyrolysis of Fe(NO3)3·9H2O on graphene sheets. The interfacial interaction between Fe3O4 and graphene nanosheets is investigated in detail by thermogravimetric and differential scanning calorimetry analysis, Raman spectrum, X-ray photoelectron energy spectrum and Fourier transform infrared spectroscopy. It was found that Fe3O4 nanoparticles disperse homogeneously on graphene sheets, and form strong covalent bond interactions (Fe–O–C bond) with graphene basal plane. The strong covalent links ensure the high specific capacity and long-period cyclic stability of Fe3O4/graphene hybrid electrodes for lithium-ion batteries at high current density. The capacity keeps as high as 796 mAhg−1 after 200 cycles without any fading in comparison with the first reversible capacity at the current density of 500 mAg−1 (ca. 0.6 C). At 1 Ag−1 (ca. 1.3 C), the reversible capacity attains ca. 550 mAhg−1 and 97% of initial capacity is maintained after 300 cycles. This work reveals an important factor affecting the high-rate and cyclic stability of metal oxide anode, and provides an effective way to the design of new anode materials for lithium-ion batteries.
Introduction
Electrode materials with the high specific capacity, high-rate performance, and long-term cyclic lifetime for next-generation lithium-ion batteries (LIBs) are in great need to supply high power for portable electronic devices and even electric vehicles.1 The transition metal oxides (such as Fe3O4, CoO, and NiO), as the potential alternative anode materials for LIBs, possess the promisingly high theoretical specific capacities, which is almost two times higher than that of carbon materials.2 However, three main drawbacks limit their application in further: (1) low electronic conductivity; (2) large volume change during the charge-discharge processes, resulting in the failure of electrical contact and structural collapse; and (3) subsequent aggregation of metal/metal oxide nanoparticles during the cycle process, due to their high surface area and activity.3–7 One of the common strategies to resolve these problems is to prepare carbon-based composite by coating metal/metal oxide with carbon layers or dispersing them into carbon matrix.3–8 It is expected that the carbon materials can, on one hand, improve the electrical conductivity of metal oxide, and on the other hand, act as a buffer for volume change and subsequent aggregation of metal oxide particles, owing to their high electronic and ionic conductivities, small volume expansion, high mechanical strength, and relative lightness. Up to now, cycle performance of metal oxide at low rate in the range from dozens to one hundred cycles can be improved efficiently in these reports.3–7 Unfortunately, the true improvement of long-period cycle performance at high rate, for most of them has not yet been achieved.The rising of graphene also opens up new opportunities for design of novel anode materials for the next-generation LIBs, because graphene nanosheets (GNSs) possess many excellent properties, including high thermal conductivity (ca. 3000 Wm−1K−1), high mechanical stiffness (1060 GPa), extraordinary electronic transport properties, and large specific surface areas (2600 m2g−1).9,10 Recently, many efforts have also been made to prepare graphene-based nanocomposites with metal oxides including SnO2,11,12Fe3O4,13–16Co3O4,17TiO2,18CuO,19 and Mn3O420 as electrode materials for LIBs. Compared with other carbon/metal oxide composites, some of graphene-based metal/metal oxide hybrids exhibited a largely improved electrochemical performance. However, their long-period cycle performance at high rate is still unsatisfactory, because their high-rate capabilities are measured using the stepwise cycles at various high current densities, and no more than 10 cycles at every current density are far from practical application (see the ESI, Table S1†). This may be attributed to that metal/metal oxide nanoparticles are still prone to aggregating into large particles during the cycles,21 because metal/metal oxide nanoparticles can only be distributed simply on the surface of graphene or between the graphene layers rather than form perfect encapsulation-structure just as shown by other carbon nanomaterials.3–7 Large volume change in the charge/discharge processes will accelerate the aggregation and further exacerbate the high-rate and cycle performances.4 One way to overcome this problem is to design elaborately the graphene-encapsulated nanostructures to segregate the metal oxide nanoparticles.21,22 For example, Yang et al.21 prepared the graphene-encapsulated Co3O4 nanoparticles. In spite of these wonderful and interesting nanostructures, their synthesis processes are also much more complex.
In addition to the design of novel nanostructures, a fundamental understanding of interfacial interaction between metal oxide and graphene is also essential to develop graphene/metal oxide anodes with excellent cyclic life and high-rate performance. The interfacial interaction between metal oxide and nanocarbon (graphene, carbon nanotubes, and even other carbon materials) in nanocarbon/metal oxide composites are associated with their preparation methods, which can be divided into in situgrowth and ex situ approach (link pre-synthesized nanoparticles to nanocarbon).23 For the latter, the interaction behaviors can be easily attributed to covalent, noncovalent, π-stacking, and electrostatic interactions, according to the pre-designed synthesis processes.23 However, for the former, which is also common method for preparation of nanocarbon/metal oxide composite anode materials,3–8,12–20 the nature of interface between metal oxide and carbon remains unclear in that a variety of chemical and physical processes for the formation of composites, and complex microstructures of most carbon materials heightened the difficulty to obtain direct information about the interface. Compared with other nanocarbons, the thin 2-D nanostructure and uniform hybridization of carbon atoms of graphene provide an ideal model to investigate in detail the interfacial interaction between metal oxide and carbon. Moreover, the interfacial interaction between metal oxide/metal and nanocarbon is of interest and also particular importance, because they not only control the catalytic growth of carbon nanotubes or graphene,24 but also play a crucial role in promoting various applications of carbon-based metal oxide composites.25,26 The interface may also determine the specific capacity and lifetime of graphene-based anode materials. In fact, enhancing or modulating the interfacial interaction between nanoparticles and nanocarbons can lead to additional novel properties25,26 as well as unique phenomena in our previous reports.7,27 Very recently, Kou et al. reported that the strong interaction between Pt nanoparticles and the ITO-graphene interface resulted in the improvement of catalytic activity of Pt-ITO-graphene.26 These efforts also inspire us to envision whether the electrochemical performance of composites can be improved largely by directly enhancing the interfacial interaction between graphene and metal/meal oxide nanoparticles. The strong interfacial interaction immobilizes closely active nanoparticles on the graphene plane during the charge/discharge process, which is expected to hinder successfully the aggregation of nanoparticles and also more efficiently take advantage of remarking properties of graphene nanosheets. However, most of the previous efforts only focus on the synthesis strategies and Li-ion storage tests of graphene-based composites. To the best of our knowledge, no investigations on the influence of interfacial interaction between metal oxides and GNS on the practical application are carried out up to now.3
In this work, we demonstrate the possibility of improving the electrochemical performance of graphene-based metal oxide by tuning the interfacial interaction between graphene and metal oxide. Using Fe3O4/graphene hybrids as an example, we probe the interfacial properties between Fe3O4 and GNS, and, for the first time, reveal the interesting influence of interfacial interaction on the electrochemical performance of the hybrid. We applied two methods to prepare Fe3O4/graphene hybrids: 1) in situ method and 2) ultrasonic mixing, and the composites obtained are named as M1-GNS and M2-GNS, respectively. This work gives new insight into the metal oxide-graphene interaction: The Fe3O4 nanoparticles in M1-GNS can be linked to graphene surface by the covalent bonding, while no distinct interaction exists in M2-GNS. When they are used as anode materials for LIBs, both M1- and M2-GNS possess similar high specific capacity and cyclic stability at a low current density. And strong interfacial interaction ensures that M1-GNS exhibits a remarkable high-rate performance: a high specific capacity of 550 mAhg−1 and excellent cyclic stability that is as long as 300 cycles without obvious fading at a current density of 1 Ag−1.
Experimental section
Preparation of graphene nanosheets
The graphene nanosheets (GNSs) were fabricated in our previous work.28 Then, the obtained GNSs were annealed at 1000 °C for 3 h in the Ar gas flow in order to remove the most oxygen-containing groups in GNSs. The GNSs after annealing were used to prepare graphene/metal oxide hybride nanosheets in the subsequent experimental procedure.Preparation of Fe3O4/graphene hybrid nanosheets
The M1-GNS was prepared by in situgrowth of Fe3O4 on GNSs surface. Typically, the GNSs (20 mg) and Fe(NO3)3·9H2O (336 mg) were dispersed in 40 ml ethanol and tip-sonicated for 30 min with a Misonix 3000 probe sonicator at 100 W. Then, after removing ethanol at 80 °C under vigorous stirring, the Fe3O4/graphene hybrid nanosheets (M1-GNS) were obtained by annealing the mixture of GNSs and Fe(NO3)3 at 500 °C for 3 h in nitrogen gas atmosphere.For comparison, the Fe3O4/graphene hybrid nanosheets (M2-GNS) with the same content of Fe3O4 as M1-GNS were also prepared by the mechanical mixing. The Fe3O4 nanoparticles were supplied by the Alfa Aesar. Typically, the GNSs (30 mg) and Fe3O4 nanoparticles (70 mg) were dispersed in 40 ml ethanol and tip-sonicated for 30 min with a Misonix 3000 probe sonicator at 100 W. After the sonication, the mixture was filtered over a PTFE membrane (0.2 μm pore size), and the filter cake was rinsed twice with 50 ml of ethanol. After washing, the Fe3O4/graphene composite was dried at 100 °C in a vacuum oven.
Characterization
Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM) measurements were carried out with a JEOL JEM-3010 F microscope operating at 300 kV. Scanning electron microscope (SEM) observation was conducted on a Hitachi S-4700 field emission scanning electron microscope.X-ray diffraction (XRD) measurements were performed with a Rigaku D/max-2500B2+/PCX system using Cu Kα radiation (λ = 1.5406 Å) over the range of 5–90° (2θ) at room temperature.
Thermogravimetry (TG) and differential scanning calorimetry (DSC) measurements were conducted on a NETZSCH STA449C simultaneous thermal instrument. The samples (ca. 5 mg) were heated from room temperature to 1000 °C at 10 °C/min under flowing oxygen (24 ml/min).
The Raman spectra were recorded from 1000 to 2000 cm−1 at room temperature using a HR 800 Raman spectrometer (produced by HORIBA Jobin Yvon company) with an excitation line of 532 nm and using an Olympus microscope and a 50 × microscopy objective to focus the laser beam onto a spot of 1 μm2.
X-ray photoelectron energy spectra (XPS) spectra were recorded using monochromatic AlK(1486.6 eV) X-ray sources with 30 eV pass energy in 0.5 eV step over an area of 650 μm × 650 μm to the sample. Before XPS measurement, the sample is degassed under a high-vacuum condition (<10−7 Pa) to remove the adsorbed water and oxygen. Atomic concentrations were calculated using peak areas of elemental lines after Shirley background subtraction and taking account of the sensitivity factors, the asymmetry parameters as well as the measured analyzer transmission function.
The functional group information of samples was measured by Fourier transform infrared (FTIR, Nicolet Nexus 670).
Brunauer-Emmett-Teller specific surface areas were measured with an ASAP 2020 Micromeritics Instrument at 77 K.
Electrochemical measurements
The electrochemical performances of the samples were tested using coin cells.29Working electrode was prepared by mixing the active mass (M1-GNS, M2-GNS, GNS, or Fe3O4), acetylene black, and poly (vinylidene difluoride) at a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. And counter electrode was the lithium sheet. Electrolyte was one molar LiPF6 solution in ethylene carbonate/dimethyl carbonate (1:1 v/v). The cells were tested at various current densities in the voltage range from 0.01 to 3.0 V. AC impedance spectra were obtained by applying a sine wave with amplitude of 5.0 mV over the frequency range from 100 kHz to 0.01 Hz on an electrochemical workstation (CHI 660B).
:10:10. And counter electrode was the lithium sheet. Electrolyte was one molar LiPF6 solution in ethylene carbonate/dimethyl carbonate (1:1 v/v). The cells were tested at various current densities in the voltage range from 0.01 to 3.0 V. AC impedance spectra were obtained by applying a sine wave with amplitude of 5.0 mV over the frequency range from 100 kHz to 0.01 Hz on an electrochemical workstation (CHI 660B).
Results and discussion
The morphologies and nanostructures of GNSs obtained were investigated by SEM and HRTEM. Fig. 1a reveals that the free-standing two-dimensional GNSs with wave-like structure are almost transparent nanosheets under the electron beam. Fig. 1b shows a representative HRTEM image of GNS, which is also not perfectly flat but exhibits many intrinsic wrinkles. The wrinkles indicate that the GNS is composed of 3 individual monoatomic graphene layers. And the interlayer spacing (ca. 3.7 Å) is also larger than that (3.354 Å) of natural graphite. XRD pattern of dried GNSs shows a broad C(002) diffraction peak at 25° (Fig. 2a), corresponding to the relative short-range order in stacked graphene sheets.28,30–32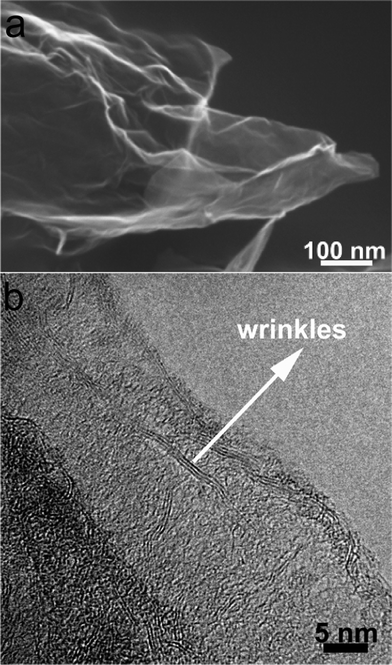 | ||
| Fig. 1 (a) SEM and (b) HRTEM images of original GNSs. | ||
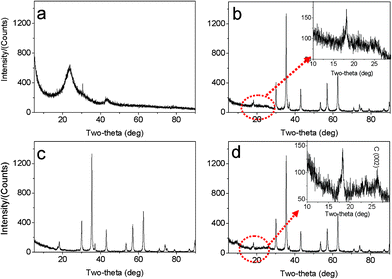 | ||
| Fig. 2 XRD patterns of (a) original GNSs, (b) M1-GNS, (c) original Fe3O4 nanoparticles, and (d) M2-GNS. | ||
The morphology and structure of M1-GNS were also investigated in detail. Its typical SEM image (Fig. 3a) shows clearly that GNS was decorated by Fe3O4 nanoparticles with the diameters in the range of 30–50 nm. The distribution of magnetite particles on graphene sheets is uniform, and no aggregated or free particles are detected. From the low-magnification TEM image (Fig. 3b), it can also be seen that all the magnetite nanoparticles supported on GNSs appear just like dark dots, and there are no free nanoparticles around GNSs. Closer observation (Fig. 3c) reveals no aggregation of nanoparticles, and morphology and size of nanoparticles are consistent with that in the SEM observations. The HRTEM image (Fig. 3d) displays the high-crystalline of nanoparticles. The lattice fringe spacing between two adjacent crystal planes of the particles was determined to be 0.29 nm, corresponding to the (220) lattice plane of cubic Fe3O4. XRD pattern of M1-GNS (Fig. 2b) also exhibits that all of the diffraction peaks are very sharp and indexed well with pure Fe3O4, indicating high crystalline structure and high phase purity of Fe3O4. Interestingly, there is no C (002) diffraction peak in the XRD pattern of M1-GNS, suggesting the disappearance of face-to-face stacking of GNSs. It is reasonable to believe that Fe3O4 nanoparticles act as a spacer to keep isolated GNSs separated according to the previous result.30 The content of Fe3O4 in M1-GNS was determined to be ca. 70 wt% by the removal of Fe3O4 in HCl solution.
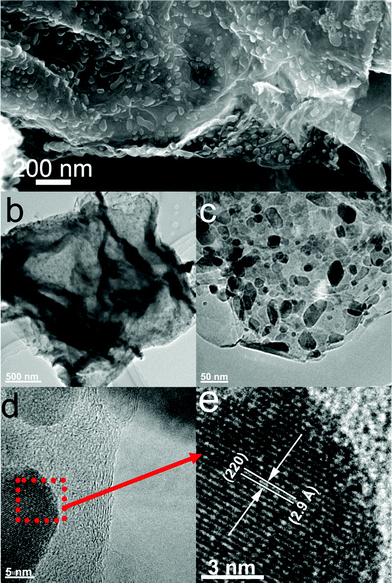 | ||
| Fig. 3 (a) SEM, (b) and (c) TEM, (d) HRTEM images of M1-GNS, and (e) enlarged HRTEM image of the selected area marked by a box in (d). | ||
For comparison, the commercial Fe3O4 nanoparticles were also employed to prepare Fe3O4/graphene hybrids (M2-GNS) with the same content as that in M1-GNS by an ultrasonic mixing method. The diffraction intensity of peaks in XRD pattern of the chosen Fe3O4 particles (Fig. 2c) is almost the same to that of M1-GNS. According to calculation by the Scherrer equation, the average diameter of commercial Fe3O4 particles is about 48.7 nm, which is slightly less than that of Fe3O4 in M1-GNS. SEM image (Fig. 4a) also reveals that size-distribution of commercial Fe3O4 is very uniform. HRTEM image of a Fe3O4 nanoparticle (Fig. 4b) confirms its high crystalline. These results suggest that the average particle size and crystallization of commercial Fe3O4 nanoparticles used here are very similar to those of the nanoparticles in M1-GNS. These magnetite nanoparticles in M2-GNS are also mixed homogeneously with GNSs. XRD pattern (Fig. 2d) shows the disappearance of C(002) peak, indicating that Fe3O4 nanoparticles are introduced uniformly to space between the isolated GNSs, and hinder successfully their aggregation due to van der Waals force. It must be mentioned that ultrasound is a very key factor to open up the interlayer between GNSs, because only a simple mechanical blending without ultrasonication (see the ESI, Fig. S1†) can not make Fe3O4 nanoparticles insert the interlayer. The SEM images of M2-GNS (Fig. 4c, d) show that Fe3O4 nanoparticles are dispersed uniformly on GNS or insert the interlayers, which is consistent with the XRD measurement (Fig. 2d). The TEM images (Fig. 4e, f) also reveal that Fe3O4 nanoparticles are loaded on the thin GNSs and no free particles around the GNSs. These results indicate that morphology and structures of M2-GNS are also very similar to those of M1-GNS. However, the interaction between Fe3O4 and GNSs in M1-GNS is much stronger than that in M2-GNS, which is confirmed by subsequent analysis.
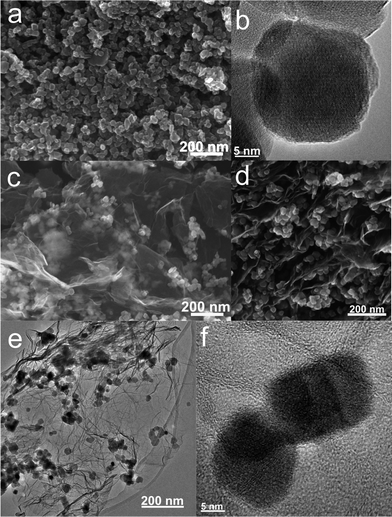 | ||
| Fig. 4 (a) SEM and (b) HRTEM images of commercial Fe3O4 nanoparticles; and (c, d) SEM, (e) TEM, and (f) HRTEM images of M2-GNS. | ||
First, the interfacial interaction in M1- and M2-GNS was investigated by TG and DSC in the oxygen atmosphere (Fig. 5), because it is well known that the types of contact (loose-or tight-contact) between the metal/metal oxide catalysts and carbons are the key factors for oxidation reactivity of carbon over the catalysts.33 For GNS, there is a dramatic mass loss at the range of 550–665 °C accompanied by an exothermic DSC peak at 650 °C, which can be attributed to the oxidation of graphene and emission of CO2/CO gas. Oxidation temperature for M2-GNS is just a little lower than that for GNS, and DSC peak is at 625 °C. However, the oxidation temperature for M1-GNS decreases largely to only 450 °C. Interestingly, the oxidation temperature of residual graphene sheets return to the 590 °C after removing Fe3O4 in M1-GNS, suggesting that Fe3O4 nanoparticles in M1-GNS accelerate the oxidation of graphene sheets. Our case is quite similar to the previous reports,33 in which Neeft et al. showed that Fe2O3 can effectively promote the oxidation of carbon in tight contact mode but perform hardly any activity in loose contact. In addition, Fig. S2 (see the ESI†) shows that the carbothermal reduction temperature (ca. 680 °C) between Fe3O4 and GNSs in M1-GNS in an inert gas atmosphere is much lower than that in M2-GNS (ca. 964 °C), further confirming the tighter contact between Fe3O4 and GNS in M1-GNS.
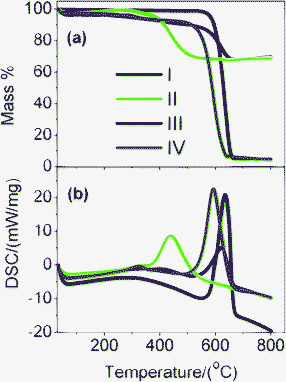 | ||
| Fig. 5 (a) TG and (b) DSC curves of (I) GNS, (II) M1-GNS, (III) M2-GNS, and (IV) graphene sheets obtained from M1-GNS after removing the Fe3O4 using HCl (1 mol/L). | ||
The different interaction behaviors between M1- and M2-GNS can also be confirmed by Raman measurement, which has been proved a powerful tool to investigate the modification of graphene and their derivatives.34Raman spectra in Fig. 6 exhibit the regular two peaks, corresponding to the D-band line (ca. 1340 cm−1) and the G-band line (ca. 1590 cm−1). G band corresponds to the first-order scattering of the E2g mode observed for sp2carbon domains, while the pronounced D band is caused by structural defects or edges that can break the symmetry and selection rule.34 The intensity ratio of D band to G band (ID/IG) is usually used to measure the graphitization degree of carbon materials. Both GNSs and M2-GNS have the same value of ID/IG (see the ESI, Table S2†), 0.93, while that of M1-GNS increases to ca. 1.04, indicating that the Fe3O4 in M1-GNS leads to the increased disorder of graphene layers. The location of G peak has ever been used to reveal the interaction between nanoparticles and graphene or carbon nanotubes.35 For GNS and M2-GNS, the G peaks locate at 1583 cm−1 (see the ESI, Table S2†). However, the G peak of M1-GNS shifts to 1592 cm−1. Generally, the shift of G peak in Raman spectra of carbon-based composite with nanocrystals means the charge transfer between carbon materials and nanocrystals.35 The Fe3O4 nanoparticles induce blue-shift of G band by 9 cm−1 in M1-GNS, suggesting the charge transfer from graphene to Fe3O4. After removing Fe3O4, the G band returns to 1585 cm−1, which should be attributed to the graphitic “self-healing”. According to the results of TG-DSC and Raman measurements, it can be concluded that there is no distinct interaction between Fe3O4 and GNS in M2-GNS, and they keep their own pristine state, while the strong interaction occurs in M1-GNS.
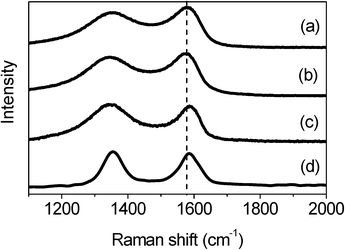 | ||
| Fig. 6 Raman spectra of (a) GNS, (b) M2-GNS, (c) M1-GNS, and (d) graphene sheets obtained from M1-GNS after removing the Fe3O4 nanoparticles. | ||
The interaction between nanocrystals and nanocarbons including graphene and carbon nanotubes can be divided into the chemical link10,36 and physical adsorption.37 The physical adsorption, especially the van der Waals force, can result in a weaker interaction, while the chemical link can lead to much stronger interaction as well as modify strongly the geometric and electronic structure of graphene due to the change of hybridization of carbon atoms from sp2 to sp3. According to analysis above, Fe3O4 in M1-GNS strongly modified the graphene plane, indicating the possible presence of chemical bonds between nanoparticles and graphene. Similar deduction has also been done in the previous reports about Fe3O4/carbon nanotubes8 and NiO/graphite,38 but what kind of bonds have not yet been identified in these researches.
It should be pointed out that the Fe3O4 nanoparticles in this case can not be possibly linked with GNSs by organic molecular chains just like shown in the previous report,39 because no surfactant was used here. The possible bonds between Fe3O4 and graphene may be: (1) that iron and carbon atoms are connected by oxygen atoms to form Fe–O–C bonds,40 and/or (2) a direct Fe–C bond.41,42XPS measurements were carried out to obtain more information. Fig. 7a shows that the GNS is composed of C and O elements, while M1-GNS is composed of C, O, and Fe elements. The curve fitting of C1s was carried out by using Gaussian-Lorentzian peak shape after a Shirley background correlation. From Fig. 7b, the C1s spectra of GNS and M1-GNS can be fitted to the mainly non-oxygenated C (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C) in aromatic rings (284.6 eV), and the C in C–O (286.1 eV) and O–CO (289.0 eV).43,44 The C–O bonds in oxygen-containing groups are dominating, which is also consistent with very recent reports.43,44 The formation of Fe–C bond can be excluded from the C1s spectrum of M1-GNS in that Fe–C bonds should be present in 283.3 eV.45 The Fe2p spectra also provide the evidence for absence of Fe–C bond. In Fig. 7c, the peak of Fe2p3/2 for M1-GNS is composed of two peaks at 710.0 and 711.7 eV, corresponding to Fe3O4.46 No peak at 707.5 eV attributed to the iron atom in Fe–C bond is presented,41 which confirms no formation of Fe–C bonds again. In addition, it can be calculated that ca. 39 at% of carbon atoms is bonded with oxygen in M1-GNS, which more by ca. 11 at% than that in the pristine GNSs (ca. 28%). It indicates the oxidation of GNSs and presence of new oxygen-containing groups during the formation of Fe3O4 nanoparticles. Therefore, it can be deduced that Fe3O4 is possibly linked with GNSs by Fe–O–C bond.
C/C–C) in aromatic rings (284.6 eV), and the C in C–O (286.1 eV) and O–CO (289.0 eV).43,44 The C–O bonds in oxygen-containing groups are dominating, which is also consistent with very recent reports.43,44 The formation of Fe–C bond can be excluded from the C1s spectrum of M1-GNS in that Fe–C bonds should be present in 283.3 eV.45 The Fe2p spectra also provide the evidence for absence of Fe–C bond. In Fig. 7c, the peak of Fe2p3/2 for M1-GNS is composed of two peaks at 710.0 and 711.7 eV, corresponding to Fe3O4.46 No peak at 707.5 eV attributed to the iron atom in Fe–C bond is presented,41 which confirms no formation of Fe–C bonds again. In addition, it can be calculated that ca. 39 at% of carbon atoms is bonded with oxygen in M1-GNS, which more by ca. 11 at% than that in the pristine GNSs (ca. 28%). It indicates the oxidation of GNSs and presence of new oxygen-containing groups during the formation of Fe3O4 nanoparticles. Therefore, it can be deduced that Fe3O4 is possibly linked with GNSs by Fe–O–C bond.
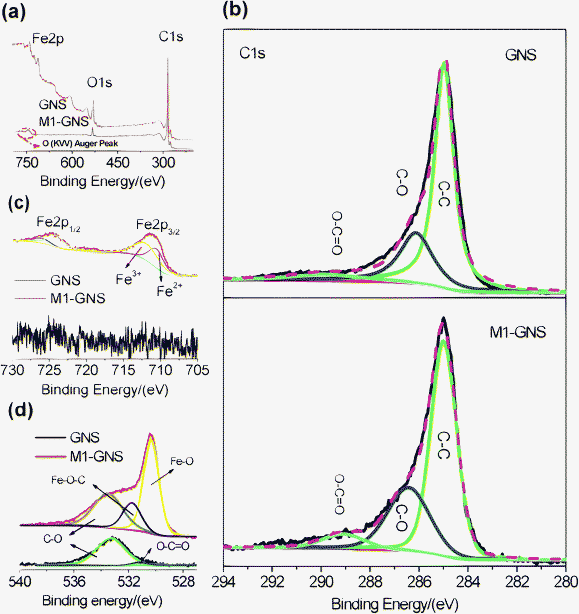 | ||
| Fig. 7 (a) XPS spectra of GNS and M1-GNS, and their (b) C1s, (c) O1s, and (d) Fe2p spectra. | ||
Furthermore, their O1s spectra are also investigated in detail, which is particularly important to confirm or disprove the existence of metal-O–C bonds.40,45 In Fig. 7d, the O1s peak of pristine GNSs is composed of two peaks centered at 533.3 and 531.2 eV. The dominating peak at 533.3 eV should be attributed to epoxy C–O groups, while the weak one at 531.2 eV corresponds to carbonyl oxygen in O–CO groups.43,44 The O1s peak of GNSs mainly comes from the residual epoxy C–O groups, which is consistent with the analysis on the C1s spectrum (Fig. 7b). The O1s peak of M1-GNS can be fitted to three peaks at 533.3, 531.7, and 530.3 eV. The peak at 533.3 eV should be attributed to the original oxygen in GNS, while one at 530.3 eV should arise from Fe3O4.46 The peak at 531.7 eV should be caused by the bonds between Fe3O4 and graphene, and/or come from the CO group, because that the binding energy of O in CO group (531.2 eV) is very close to this peak. However, this peak disappears in the residual GNS after Fe3O4 is removed (see the ESI, Fig. S3†), and is also not detected in M2-GNS (see the ESI, Fig. S3†). Therefore, the possibility, that the peak comes mainly from CO, is ruled out. Thus, the peak at 531.7 eV in M1-GNS should be attributed mainly to the bond of Fe–O–C formed between graphene and Fe3O4, which can also be confirmed from the results in previous reports40 that the binding energy of O1s in Fe–O–C bond can present in the range of 531–533 eV. Additional evidences supporting the formation of Fe–O–C bond are O1s peaks for other metal-O–C bonds including Cu–O–C, Ag–O–C, and Zr–O–C,45 in which the O1s peak can shift positively by ca. 1–3 eV than that in metal-O bonds (see the ESI, Table S3†).
FTIR spectra also support the existence of Fe–O–C bonds in M1-GNS. Fig. 8a shows that three peaks present in the FTIR spectrum of GNS. The peak located at 1733 cm−1 is related to the CO stretching.44,47 The next one presented at 1560 cm−1 should be attributed to the ring vibrations throughout the carbon skeleton, while the peak at 1222 cm−1 should be caused by epoxy (C–O–C) groups.47 In comparison with GNS, M1-GNS exhibits obviously two new IR peaks (Fig. 8b). One peak at 567 cm−1 should be attributed to the Fe–O stretching in Fe3O4. The other is a strong wide absorption peak centered at 1110 cm−1, which should be associated with the stretching of C–O in graphene.47 Interestingly, the intensity of peak at 1110 cm−1 decreases obviously as Fe3O4 nanoparticles are removed (Fig. 8c), revealing that C–O functional groups are also linked to the Fe3O4 nanoparticles. According to the previous report,48 in which the vibration of C–O in Fe–O–C bond is at about 1090 cm−1, it can also identify that the presence of the new wide peak at 1110 cm−1 also indicates the formation of Fe–O–C bonds in M1-GNS.
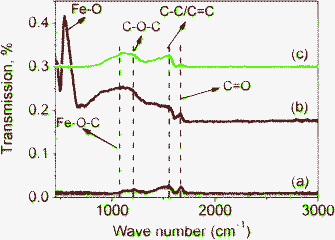 | ||
| Fig. 8 FTIR transmittance spectra of (a) GNS, (b) M1-GNS and (c) residual graphene sheets from M1-GNS after removing Fe3O4 nanoparticles. | ||
Subsequently, the electrochemical performance of M1- and M2-GNS as well as GNS and Fe3O4 nanoparticles were evaluated by galvanostatic charge/discharge measurements. Previous reports have also revealed that the electrochemical performance of metal oxide anode materials was influenced by many factors, including size, morphology, specific surface area, dispersion of oxide in carbon matrix, etc.2,8 According to the previous analysis, morphology, size, and dispersion of Fe3O4 nanoparticles in M1- and M2-GNSs are very alike. The specific surface area of M2-GNSs (150 m2/g) is larger than that of M1-GNS (96 m2/g), which also should be attributed to the loose contact between Fe3O4 nanoparticles and GNSs resulting in more pores in M2-GNS. Therefore, significant difference between M2-GNS and M1-GNS is that the interfacial interaction in M1-GNS is much stronger than that in M2-GNS. This obvious difference should also lead to the difference of their electrochemical performances as anode materials for LIBs.
The GNS shows the higher specific capacity and better cyclic stability. The first discharge capacity and reversible capacity of GNS are 1928 and 753 mAhg−1 (Fig. 9a), respectively. The specific capacity decreases gradually to ca. 550 mAhg−1 after 10 cycles and keeps it up at subsequent 40 cycles (Fig. 9d). The theoretical specific capacity of Fe3O4-graphene composites with 30 wt% graphene is ca. 873 mAhg−1 calculated from the theoretic capacity of Fe3O4 (924 mAhg−1) and the first reversible capacity of GNS (753 mAhg−1). The first discharge and charge capacities of Fe3O4 nanoparticles are 1272 and 951 mAhg−1, respectively. However, the capacity fades quickly, and only 112 mAhg−1 remains after 50 cycles, indicating the poor cycle performance.
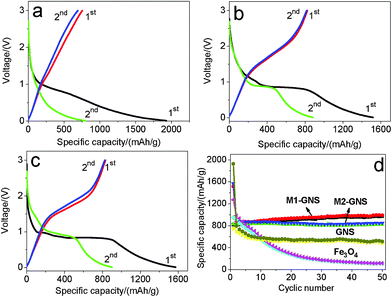 | ||
| Fig. 9 The initial two charge/discharge curves of (a) original GNS, (b) M1-GNS and (c) M2-GNS, and (d) cycling performance of M1-GNS, M2-GNS, GNS, and Fe3O4 nanoparticles at the current density of 50 mAg−1. | ||
After mixing with GNSs, M1-GNS electrode exhibits high cyclic stability at a current density of 50 mAg−1. Fig. 9b shows its initial two charge-discharge profiles. The first discharge capacity and reversible capacity are 1516 and 825 mAhg−1, respectively. The specific capacity shows a slight increase at the subsequent cycles. The reversible capacity increases stably to ca. 951 mAhg−1 without any capacity fading after 50 cycles (Fig. 9d).
M2-GNS electrode also exhibits high cyclic stability at 50 mAg−1, although it was prepared only through the simple ultrasonic mixing. The first discharge capacity and reversible capacity are 1572 and 832 mAhg−1 (Fig. 9c), respectively. The higher first discharge capacity (1572 mAhg−1) should be related with the higher specific surface area of M2-GNS. After 50 cycles the reversible capacity still maintains as high as 826 mAhg−1, which is ca. 99% of the first reversible capacity. Therefore, at low current density M1-GNS has not exhibited more significant advantages in cycle performance compared with M2-GNS.
It should be noted that a capacity rise with cycles occurs in M1-GNS electrode. This phenomenon had been observed by other researchers on carbon/iron oxide49 and Cu nanowires/Fe3O4 anode materials,50 and was attributed to the formation of gel-like films caused by decomposition of electrolyte.49 However, it does not present in all of the C/iron oxide anode systems,6,7 including our M2-GNS, so the capacity increase should also be related with the interfacial interaction between carbon (or Cu) and iron oxide. The strong interfacial interaction in M1-GNS possibly promotes the quick transfer of electron between graphene and Fe3O4 to some extent as shown below, and leads to the slight increase of specific capacity with cycle. Further research is proceeding.
We also examined the electrochemical performance of the Fe3O4/GNSs composite prepared by only a simple mechanical blending without ultrasonication (see the ESI, Fig. S1†). Its reversible capacity for the first cycle is ca. 825 mAhg−1, and fades gradually to 428 mAhg−1 after 50 cycles. The cyclic stability is improved compared with that of the pure Fe3O4 nanoparticles, but much lower than those of M1- and M2-GNS, which should arise from that the aggregation of graphene layers due to very strong van der Waals interactions decrease the flexibility and connectivity of graphene network.8 This also indicates the homogenous mixing between Fe3O4 and GNSs has an important effect on the cycle performance of graphene-based anode at low rate.
In further, the electrochemical performance of M2-GNS was measured at higher current densities. Fig. 10a shows that the first discharge and reversible capacities are ca. 1339 and 730 mAhg−1, respectively, at 500 mAg−1 (ca. 0.6 C), possessing ca. 88% of capacities at 50 mAg−1. The reversible capacity decreases gradually to 512 mAhg−1 after 50 cycles (Fig. 10c). Even so, this value is comparable to and even superior to those of other carbon/Fe3O4 nanoparticles reported previously.6,7 Considering the simple preparation method, it confirms the charming properties of graphene once again. At 1 Ag−1, the capacity fading of M2-GNS becomes more obvious. The first discharge capacity and reversible capacity are ca. 977 and 574 mAhg−1 (Fig. 10a), respectively, but the capacity decrease quickly to only 119 mAhg−1 after 50 cycles (Fig. 10d).
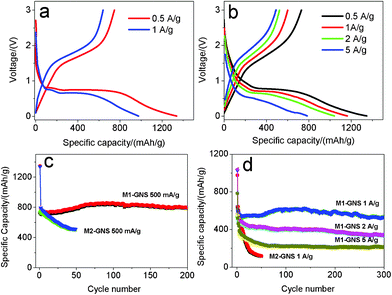 | ||
| Fig. 10 The first charge/discharge curves of (a) M2-GNS, and (b) M1-GNS, and (c, d) corresponding their cycling performance at various current densities. | ||
The most important advantage for M1-GNS is the outstanding high-rate performance compared with M2-GNS. At 500 mAg−1, the values of the first discharge (1346 mAhg−1) and reversible capacity (730 mAhg−1) of M1-GNS are also almost the same with those of M2-GNS (Fig. 10b). However, the cyclic stability of M1-GNS is much better than that of M2-GNS. The capacity of M1-GNS after 200 cycles is still up to 796 mAhg−1 without any fading (Fig. 10c). At 1 Ag−1, the reversible capacity attains ca. 550 mAhg−1 and still keeps 531 mAhg−1 even after a long cycle period of 300 cycles (Fig. 10d). At the higher current density, the reversible capacity of M1-GNS decreases, but the cyclic stability remains very well. The first discharge and reversible capacity of M1-GNS is up to 1040 and 523 mAhg−1 even at 2 Ag−1 (ca. 2.3 C), and the capacity keeps at 335 mAhg−1 after 300 cycles (Fig. 10d). It can be calculated that the capacity fading is only ca. 0.6 mAhg−1 per cycle at ca. 2.3 C. When the current density is further improved to 5 Ag−1 (ca. 5.7 C), the reversible capacity attains 491 mAhg−1 and remains 213 mAhg−1 after 300 cycles (Fig. 10d). And the capacity fading is still less than 1 mAhg−1 for one cycle.
It needs to be pointed out that both the specific capacity and cyclic stability of M1-GNS at high rate are also superior to the reported values of other carbon-based Fe3O4 composites,6,7 and even graphene/Fe3O4 hybrids (See the ESI, Table S1†).13–16 For example, very recently, Wang et al.13 reported that the Fe3O4/graphene nanocomposite with a graphene content of 38.0 wt% exhibits a capacity of ca. 650 mAhg−1 at 100 mAg−1 after the 100 cycles. Zhou et al.14 reported that graphene-wrapped Fe3O4 anode materials show a reversible capacity of ca. 580 mAhg−1 at 700 mAg−1 after 100 cycles. Zhang et al.15 reported that Fe3O4/graphene composite exhibits a high initial reversible capacity of 1030 mAhg−1 at 0.1 C, but their capacity decreases to ca. 650 mAhg−1 after 50 cycles. Indeed, these initial efforts have improved obviously the electrochemical performance of Fe3O4, but the reported results are only close to or a little higher than that of M2-GNS, and lower than that of M1-GNS, especially at high current density. This may be attributed mainly to the weak interfacial interaction between Fe3O4 and graphene sheets in these reports. It is worthy to note that their main preparation procedures of Fe3O4/graphene composites were in the liquid phase, where the lower reaction temperature may lead to the difficulty of forming strongly covalent-bond link between nanoparticles and graphene sheets.
To reveal in further the reasons for excellent high-rate performance of M1-GNS, the kinetics of both M1- and M2-GNS electrodes were investigated by the electrochemical impedance spectroscopy measurement (see the ESI, Fig. S4†). By simulation, it can be found that the film resistance (Rf) and charge-transfer resistance (Rct) of M2-GNS are ca. 5.2 and 11.5 Ω, respectively. Both the Rf (3.6 Ω) and Rct (6.8 Ω) of M1-GNS are much lower than those of M2-GNS electrode, indicating the formation of a better conductive network in M1-GNS. This should be attributed to the stronger interaction between Fe3O4 nanoparticles and GNS in M1-GNS. The charge-transfer resistance in Fe3O4/graphene hybrids involves the resistance in the graphene basal plane (Rg) as well as that between Fe3O4 and graphene sheets (RFeO-g). The Fe–O–C covalent link between Fe3O4 nanoparticles and GNS in M1-GNS can lead to the increase of Rg in that the formation of covalent interrupts the large sp2 domain sizes.43 However, the covalent bond should decrease the RFeO-g, because the charge transfer from graphene to Fe3O4 takes place as shown in Raman spectrum (Fig. 6). The coupling of the two factors eventually results in the higher conductivity of M1-GNS than that of M2-GNS, so the electron transport between Fe3O4 and graphene sheets is a limiting step. In addition, because of the strong interaction, the active nanoparticles after cycles still tightly and homogenously located at graphene sheets in spite of their volume expansion (see the ESI, Fig. S5†).
Based on the analysis above, it is concluded that the electrochemical performance of graphene-based composites with metal oxide can be improved largely by adjusting the interaction between graphene and metal oxide. In fact, the contact types of nanocrystals to graphene nanosheets include the physisorption, electrostatic binding, covalent bond and charge transfer interactions. The exploration of these acting forces in oxide electrodes would be beneficial for the improvement of the stability and rate performance, and the design of new electrode material systems, which will be necessary for the next research. In addition, synthesis method of M1-GNS also possesses several striking merits by compared with previous reports. First, the synthesis process is very simple, and is easy to be applied in large-scale preparation. Second, this process is eco-friendly and low-cost in that no expensive and toxic organic metals and solvents are used. Third, this method should also be extended easily to other metal oxides (e.g.: CoO, NiO, and CuO).
Conclusions
In this work, we prepared Fe3O4/graphene-nanosheets composites by a facile in situ method, and showed experimentally that Fe3O4 nanoparticles were contacted with graphene nanosheets through the Fe–O–C bonds. For the first time, it was confirmed that the strongly interfacial interaction endows the enhancement of the electrical performance. The Fe3O4/GNS hybrid nanosheets exhibit the excellent high-rate performance of 796 mAhg−1 after 200 cycles at the current density of 500 mAg−1 (ca. 0.6 C) and 531 mAhg−1 after 300 cycles at 1 Ag−1 (ca. 1.3 C). Our work provides a detailed knowledge on the interfacial interaction between metal oxide and graphene, points to a new key factor affecting the lithium-ion batteries, and also opens up a suitable strategy to improve the electrochemical performances of metal oxide anode. This fundamental strategy should also be applicable to improve the electrochemical performance of anode materials such as Co3O4, CuO and SnO2. In addition, it is also promising to consider the influence of interfacial interaction on the application of graphene/nanocrystal hybrids to other fields, such as nanoelectronics, sensing, catalysis, fuel cells, solar cells, and supercapacitors.Acknowledgements
This work was supported by the National Natural Science Foundation of China (50572003 and 50972004), the Opening Project of Xinjiang Key Laboratory of Electronic Information Materials and Devices (XJYS0901-2010-03), and Foundation of Excellent Doctoral Dissertation of Beijing City (YB20081001001).References
- (a) P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS; (b) D. S. Su and R. Schlögl, ChemSusChem, 2010, 3, 136 CrossRef CAS; (c) M. Pumera, Energy Environ. Sci., 2011, 4, 668 RSC; (d) Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1114 Search PubMed.
- (a) P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS; (b) S. Grugeon, S. Laruelle, R. Herrera-Urbina, L. Dupont, P. Poizot and J.-M. Tarascon, J. Electrochem. Soc., 2001, 148, A285 CrossRef CAS; (c) Y. NuLi, R. Zeng, P. Zhang, Z. Guo and H. Liu, J. Power Sources, 2008, 184, 456 CrossRef CAS.
- (a) J. Hassoun, G. Derrien, S. Panero and B. A. Scrosati, Adv. Mater., 2008, 20, 3169 CrossRef CAS.
- (a) G. Cui, Y.-S. Hu, L. Zhi, D. Wu, I. Lieberwirth, J. Maier and K. Müllen, Small, 2007, 3, 2066 CrossRef CAS; (b) K. T. Lee, Y. S. Jun and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652 CrossRef CAS.
- Y. Yu, L. Gu, C. Wang, A. Dhanabalan, P. A. Aken and J. Maier, Angew. Chem., Int. Ed., 2009, 48, 6485 CrossRef CAS.
- (a) W.-M. Zhang, X.-L. Wu, J.-S. Hu, Y.-G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941 CrossRef CAS; (b) H. Liu, G. Wang, J. Wang and D. Wexler, Electrochem. Commun., 2008, 10, 1879 CrossRef CAS.
- J. Zhou, H. Song, X. Chen, L. Zhi, S. Yang, J. Huo and W. Yang, Chem. Mater., 2009, 21, 2935 CrossRef CAS.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22, E145 CrossRef CAS.
- (a) K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS; (b) M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. M. Blake, P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72 CrossRef CAS.
- (a) G. Wang, B. Wang, X. Wang, J. Park, S. Dou, H. Ahn and K. Kim, J. Mater. Chem., 2009, 19, 8378 RSC; (b) L.-S. Zhang, L.-Y. Jiang, H.-J. Yan, W. D. Wang, W. Wang, W.-G. Song, Y.-G. Guo and L.-J. Wan, J. Mater. Chem., 2010, 20, 5462 RSC.
- J.-Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z.-X. Wang, L.-Q. Chen and H.-K. Liu, Chem.–Eur. J., 2011, 17, 661 CrossRef CAS.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Chen, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- M. Zhang, D. Lei, X. Yin, L. Chen, Q. Li, Y. Wang and T. Wang, J. Mater. Chem., 2010, 20, 5538 RSC.
- (a) P. Lian, X. Zhu, H. Xiang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 56, 834 CrossRef CAS; (b) B. Li, H. Cao, J. Shao, M. Qu and J. H. Warner, J. Mater. Chem., 2011, 21, 5069 RSC.
- (a) S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236 CrossRef CAS; (b) Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 6, 3187 CrossRef; (c) S. Q. Chen and Y. Wang, J. Mater. Chem., 2010, 20, 9735 RSC.
- (a) D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS; (b) Y. Qiu, K. Yan, S. Yang, L. Jin, H. Deng and W. Li, ACS Nano, 2010, 4, 6515 CrossRef CAS.
- B. Wang, X.-L. Wu, C.-Y. Shu, Y.-G. Guo and C.-R. Wang, J. Mater. Chem., 2010, 20, 10661 RSC.
- H. Wang, L.-F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408 CrossRef CAS.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587 CrossRef CAS.
- (a) V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679 RSC; (b) D. Eder, Chem. Rev., 2010, 110, 1348 CrossRef CAS; (c) F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139 CrossRef CAS.
- (a) H. Yoshida, S. Takeda, T. Uchiyama, H. Kohno and Y. Homma, Nano Lett., 2008, 8, 2082 CrossRef CAS; (b) B. Wang, X. Ma, M. Caffio, R. Schaub and W.-X. Li, Nano Lett., 2011, 11, 424 Search PubMed.
- (a) F. Banhart, Nanoscale, 2009, 1, 201 RSC; (b) D. Eder, Chem. Rev., 2010, 110, 1348 CrossRef CAS; (c) G. Hong, S. M. Tabakman, K. Welsher, H. Wang, X. Wang and H. Dai, J. Am. Chem. Soc., 2010, 132, 15920 CrossRef CAS.
- R. Kou, Y. Shao, D. Mei, Z. Nie, D. Wang, C. Wang, V. Viswanathan, S. Park, I. A. Aksay, Y. Lin, Y. Wang and J. Liu, J. Am. Chem. Soc., 2011, 133, 2541 CrossRef CAS.
- (a) J. Zhou, H. Song, X. Chen and J. Huo, J. Am. Chem. Soc., 2010, 132, 11402 Search PubMed; (b) J. Zhou, H. Song, X. Chen, L. Zhi, J. Huo and B. Cheng, Chem. Mater., 2009, 21, 3730 CrossRef CAS.
- P. Guo, H. Song and X. Chen, Electrochem. Commun., 2009, 11, 1320 CrossRef CAS.
- (a) S. Yang, J. Huo, H. Song and X. Chen, Electrochem. Commun., 2006, 8, 137 CrossRef CAS; (b) J. Zhou, H. Song, B. Fu, B. Wu and X. Chen, J. Mater. Chem., 2010, 20, 2794 RSC.
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792 CrossRef CAS.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845 CrossRef CAS.
- X. Du, P. Guo, H. Song and X. Chen, Electrochim. Acta, 2010, 55, 4812 CrossRef CAS.
- (a) J. P. A. Neeft, M. Makkee and J. A. Moulijin, Chem. Eng. J., 1996, 64, 295 Search PubMed; (b) J. P. A. Neeft, M. Makkee and J. A. Moulijin, Appl. Catal., B, 1996, 8, 57 Search PubMed.
- (a) K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS; (b) S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau, W. A. deHeer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061 CrossRef CAS; (c) M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751 CrossRef CAS.
- (a) A. M. Rao, P. C. Eklund, S. Bandow, A. Thess and R. E. Smalley, Nature, 1997, 388, 257 CrossRef CAS; (b) K. S. Subrahmanyam, A. K. Manna, S. K. Pati and C. N. R. Rao, Chem. Phys. Lett., 2010, 497, 70 CrossRef CAS; (c) R. Kitaura, N. Imazu, K. Kobayashi and H. Shinohara, Nano Lett., 2008, 8, 693 CrossRef CAS.
- (a) S.-W. Kim, D.-H. Seo, H. Gwon, J. Kim and K. Kang, Adv. Mater., 2010, 22, 5260 Search PubMed; (b) J. A. Rodríguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422 CrossRef CAS.
- G. Lu, S. Mao, S. Park, R. S. Ruoff and J. Chen, Nano Res., 2009, 2, 192 CrossRef CAS.
- S. K. Sharma, F. J. Vastola and P. L. Valker Jr, Carbon, 1996, 34, 1407 Search PubMed.
- F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139 CrossRef CAS.
- (a) G. Kataby, M. Cojocaru, R. Prozorov and A. Gedanken, Langmuir, 1999, 15, 1703 CrossRef CAS; (b) C. Combellas, M. Delamar, F. Kanoufi, J. Pinson and F. I. Podvorica, Chem. Mater., 2005, 17, 3968 CrossRef CAS.
- H. Li, T. Xu, C. Wang, J. Chen, H. Zhou and H. Liu, Tribol. Lett., 2005, 19, 231 Search PubMed.
- A. Adenier, M.-C. Bernard, M. M. Chehimi, E. Cabet-Deliry, B. Desbat, O. Fagebaume, J. Pinson and F. Podvorica, J. Am. Chem. Soc., 2001, 123, 4541 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli and S. Miller, Adv. Funct. Mater., 2009, 19, 2577 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhovalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581 CrossRef CAS.
- (a) B. L. Hurley and R. L. McCreery, J. Electrochem. Soc., 2004, 151, B252 CrossRef CAS; (b) S. Serghini-Monim, P. R. Norton, R. J. Puddephatt, K. D. Pollard and J. R. Rasmussen, J. Phys. Chem. B, 1998, 102, 1450 CrossRef CAS; (c) C. Dicke, M. Morstein and G. Hahner, Langmuir, 2002, 18, 336 Search PubMed.
- P. C. J. Graat and M. A. J. Somers, Appl. Surf. Sci., 1996, 100/101, 36 CrossRef.
- M. Acik, G. Lee, C. Mattervi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840 CrossRef CAS.
- H. Peng, X. Zhang, K. Huang and H. Xu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2008, 23, 480 Search PubMed.
- (a) L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, J. Power Sources, 2008, 183, 717 CrossRef CAS; (b) P. Adelhelm, Y.-S. Hu, M. Antonietti, J. Maier and B. M. Smarsly, J. Mater. Chem., 2009, 19, 1616 RSC.
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J.-M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image, XRD pattern, and electrochemical performance of Fe3O4/graphene composite prepared by simple mechanical blending without ultrasonication, TG-DSC curves of M1-GNS and M2-GNS at the Ar atmosphere, XPS spectrum of O1s in graphene sheets obtained after removing the Fe3O4 from M1-GNS and M2-GNS, SEM and TEM images of M1-GNS after discharge-charge cycles, tables of electrochemical performance of graphene/metal oxide composites reported in the literatures, data for Raman spectra, binding energy of O1s in various bonds, Electrochemical Impedance Spectra of M1-GNS and M2-GNS. See DOI: 10.1039/c1ra00402f |
| This journal is © The Royal Society of Chemistry 2011 |