A novel stereoselective one-pot synthesis of 2-susbstituted amino-5,6-dihydro-4H-1,3-thiazines via primary allylamines afforded from Morita–Baylis–Hillman acetates†‡
Subhendu
Bhowmik§
,
Amita
Mishra§
and
Sanjay
Batra§
*
Medicinal and Process Chemistry Division, CSIR-Central Drug Research Institute (CSIR), PO Box 173, Lucknow, 226001, India. E-mail: batra_san@yahoo.co.uk; s_batra@cdri.res.in; Fax: +91-522-2623405; Tel: +91-522-2621411-18 Extn. 4234, 4368
First published on 16th September 2011
Abstract
A new stereoselective synthesis of 2-susbstituted amino-5,6-dihydro 4H-1,3-thiazines using primary allylamines obtained from the Morita–Baylis–Hillman (MBH) acetates is described. The primary allylamines react with aryl isothiocyanates to afford the title compounds via sequential thiourea formation and intramolecular sulfa-Michael reaction in a one-pot process under two different experimental conditions. Two steps during the reactions between the allylamines derived from the MBH adducts of benzaldehyde or electron-donating group bearing substituted benzaldehydes and aryl isothiocyanates proceed in a cascade sequence to directly afford the anti-isomer of the title compounds. In contrast reactions between the allylamines generated from the MBH adducts of electron-withdrawing group containing substituted benzaldehydes and aryl isothiocyanate result in allyl thioureas which undergo Lewis acid-mediated intramolecular sulfa-Michael cyclization to afford syn or anti-products depending on the placement of the substitution on the phenyl ring. A plausible mechanism is proposed to explain the observed stereoselectivity amongst the prepared 1,3-thiazines.
Introduction
5,6-Dihydro-1,3-thiazine represents an important structural motif which is core of biologically active natural product cephamycin, antibiotic cephalosporin and several other compounds displaying a variety of bioactivities (Fig. 1).1 In particular, compounds bearing this scaffold are useful as insecticides, agrochemicals, medicinal microbicides and cannabinoid receptor antagonists.2 However there are only a few methods available for the synthesis of this core unit.3 For example, the Diels Alder reaction of 3-aza-4-dimethylamino-l-thia-butadienes with acrylates under heating or by application of high pressure furnish 5,6-dihydro-4H-1,3-thiazines in good yields.4 On the other hand MgBr2-promoted Diels Alder reaction of diene with chiral N-acryloyl-2-oxazolidinone derivative followed by removal of chiral auxiliary successfully produced chiral substituted 5,6-dihydro-4H-1,3-thiazines.5 Dallemange and co-workers have successfully achieved the synthesis of this scaffold from readily available aryl-amino acids.6 We herein describe a new straightforward stereoselective approach to 5,6-dihydro-4H-1,3-thiazines from the primary allylamines derived from the Morita–Baylis–Hillman (MBH) acetates. | ||
| Fig. 1 Core structure of Cephamycin and Cephalosporin. | ||
The propensity of the MBH reaction to afford multifunctional products which have the ability to undergo an array of chemical transformations has increased its utility manifold in synthetic organic chemistry.7 In particular, the products easily accessible via nucleophilic substitutions in MBH adducts or acetates are focal intermediates for synthesis of a plethora of cyclic frameworks employing robust protocols.7d Where as predominantly C-, O- or N-centred nucleophiles have been used resulting in either carbocycles, oxacycles or aza-cycles, only a few reactions of MBH derivatives with sulfur-nucleophiles leading to sulfur-containing cyclic compounds have been reported. Sa et al. reported the synthesis of 1,3-thiazinones from allylic thiocyanates whereas Yadav et al. achieved the synthesis of substituted 2-thioxo-2H-1,3-thiazin-4(3H)-ones and 4-imino-3,4-dihydro-2H-1,3-thiazine-2-thiones from the allylic dithiocarbamates.8,9 It may be noted that preparation of these compounds was accomplished via initial sulfa-Michael reaction followed by intramolecular amide coupling leading to cyclization. Recently we have disclosed a benign synthesis of primary allylamines from the MBH acetates using aqueous ammonia and demonstrated their application for the construction of tetrazole-fused pyrimidinones.10 In a continuing program of our research group to develop general protocols for affording heterocyclic compounds from the MBH derivatives, we reasoned that such primary allylamines could be a viable precursor to substituted 5,6-dihydro-4H-1,3-thiazines. In principle, a reaction between the allylamine and arylisothiocyanate would afford allyl thioureaII, which could subsequently undergo an intramolecular cyclizationvia sulfa-Michael reaction11 to produce the envisaged 1,3-thiazineIV (Scheme 1). Based on our prior experience with substituted allylamines however, the isomerisation reaction of allyl thiourea (II) to produce product III is not ruled out.12 During the course of this study Chen and Clive reported a general synthesis of thiazepinesvia sulfa-Michael reaction in the MBH derivatives which bears close analogy to our envisaged approach.13
 | ||
| Scheme 1 Retrosynthetic plan for functionalized 1,3-thiazinesviathiourea. | ||
The study commenced with synthesis of the primary allylamines (2) from MBH acetates 1. The change of solvent in the reported procedure10 from a mixture of THF:H2O to methylene chloride:H2O in the conversion of MBH acetates (1) to allylamines (2) produced better yields of the amines which could be easily isolated and employed directly for further reactions (Scheme 2). This modified procedure was adopted for preparing all amines2.1a–g and 2.2a–h derived from MBH acetates of acrylates and acrylonitrile, respectively. In order to examine the feasibility of the approach initial optimization studies were performed with the amine2.1a. In the first stage the crude amine2.1a was treated with phenyl isothiocyanate (A) in methylene chloride at room temperature (Table 1). The reaction was observed to be complete in 6 h to afford a product in 61% yields. Based on the spectral analysis, this product was established to be methyl 4-phenyl-2-(phenylamino)-5,6-dihydro-4H-1,3-thiazine-5-carboxylate (3.1aA). It was pleasing to note that the reaction was remarkably stereoselective to afford the anti isomer exclusively. The stereochemical assignment of anti-3.1aA was based on the observed 7.7 Hz coupling constants for the protons present at C–5 and C–6.14 Under the optimized conditions the scope of the protocol was explored using a variety of substrates. In the first set of experiments, 2.1a and 2.2a were treated with different substituted phenyl isothiocyanates (A–C) in methylene chloride at room temperature. The reactions were found to be complete in 6 h to stereoselectively afford the anti isomer of the respective 2-susbstituted amino-5,6-dihydro-4H-1,3-thiazines (anti-3.1aB–C,anti-3.2aA–C) in 60–72% yields (Table 1).
 | ||
| Scheme 2 The synthesis of allylamines. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Allylamine | EWG | Ar (A–C) | Product (anti only) | Yield (%)a |
| a Isolated yields after column chromatography. | |||||
| 1 | 2.1a | CO2Me | Ph (A) | 3.1aA | 61 |
| 2 | 2.1a | CO2Me | 4-ClC6H4 (B) | 3.1aB | 65 |
| 3 | 2.1a | CO2Me | 2-BrC6H4 (C) | 3.1aC | 60 |
| 4 | 2.2a | CN | Ph (A) | 3.2aA | 64 |
| 5 | 2.2a | CN | 4-ClC6H4 (B) | 3.2aB | 72 |
| 6 | 2.2a | CN | 2-BrC6H4 (C) | 3.2aC | 65 |
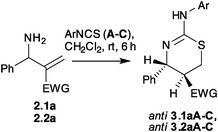
Next, scope of the protocol described in Table 1, was extended by reacting diverse allylamines with phenyl isothiocyanate (A). Hence 2.1b–g and 2.2b–h were treated with A in methylene chloride at room temperature. In general the reactions were found to be complete in 3–5 h. From the analysis of the spectroscopic data of the products, we were intrigued to note that only the allylamines (2.1b, 2.2b,h) containing a phenyl ring bearing electron donating groups such as methyl or methoxy produced the expected 5,6-dihydro-1,3-thiazines (anti-3.1bA and anti-3.2b,hA). Unlike, the allylamines (2.1c–g and 2.2c–e,g) possessing the phenyl ring substituted with electron withdrawing substituents such as halo groups furnished the corresponding allyl thiourea derivatives 4.1c–gA and 4.2c–e,gA exclusively (Table 2). Against the trend 2.2f, however afforded the respective 1,3-thiazine (anti-3.2fA) directly under similar reaction conditions. In our attempt to accomplish the intramolecular sulfa-Michael reaction in 4, altered conditions such as prolonging the reaction time or heating the thiourea at different temperatures were investigated, but the desired objective could not be realized. Essentially to find a condition which may induce the desired intramolecular sulfa-Michael in thiourea 4 we screened the literature. We came across several reports describing the use of Lewis acids or organo-catalysts to induce such reaction.11 In a representative study therefore, thiourea4.1cA was treated with InCl3 and L-proline separately. To our delight, the reaction of 4.1cA with InCl3 in methylene chloride at room temperature for 20 h resulted in a single product that was spectroscopically analyzed to be the required 5,6-dihydro-4H-1,3-thiazine (3.1cA). In contrast to previous results however, the stereochemistry of the isolated 3.1cA was established to be syn. On the other hand the attempted L-proline-mediated sulfa-Michael reaction of 4.1cA was unsuccessful. In a subsequent investigation to examine the competence of other Lewis-acids to promote the sulfa-Michael reaction, we discovered that In(OTf)3 and Sc(OTf)3 were also effective. Since the thiourea formation and InCl3-promoted sulfa-Michael reaction employed methylene chloride as the medium, we were tempted to explore the possibility of accomplishing the two steps in one-flask as a one-pot procedure. Thus, amine2.1c was treated with A at room temperature and as soon as the starting material was consumed (ca 30 min), InCl3 was added to the reaction mixture. The reaction was complete in 20 h to yield a single product identified to be the syn-3.1cA with improved yield. In view of this result, the approach was investigated using other allylamines which have produced thiourea. Therefore allylamines 2.1d–g and 2.2c–e,g were initially reacted with A followed by the addition of InCl3. It was satisfying to note that these reactions resulted in the corresponding 5,6-dihydro-1,3-thiazines 3.1d–gA and 3.2c–e,gA. But it was unexpected to discover that though thiazines3.1c–eA and 3.2c–eA were isolated as syn-isomers, thiazines3.1f–gA and 3.2gA were produced as anti-isomers (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | EWG | 2 | 4 (yield %) | 3 (yield %)a |
| a Isolated yields after column chromatography. b Yields are from two-step one-pot reaction. | |||||
| 1 | 4-MeC6H4 | CO2Me | 2.1b | — | anti-3.1bA (61) |
| 2 | 4-MeC6H4 | CN | 2.2b | — | anti-3.2bA (64) |
| 3 | 2-BrC6H4 | CO2Me | 2.1c | 4.1cA (76) | syn-3.1cA (77)b |
| 4 | 2-BrC6H4 | CN | 2.2c | 4.2cA (73) | syn-3.2cA (77)b |
| 5 | 2-ClC6H4 | CO2Me | 2.1d | 4.1dA (69) | syn-3.1dA (68)b |
| 6 | 2-ClC6H4 | CN | 2.2d | 4.2dA (77) | syn-3.2dA (80)b |
| 7 | 2-FC6H4 | CO2Me | 2.1e | 4.1eA (85) | syn-3.1eA (86)b |
| 8 | 2-FC6H4 | CN | 2.2e | 4.2eA (80) | syn-3.2eA (84)b |
| 9 | 4-ClC6H4 | CO2Me | 2.1f | 4.1fA (79) | anti-3.1fA (71)b |
| 10 | 4-ClC6H4 | CN | 2.2f | — | anti-3.2fA (64) |
| 11 | 3,4-Cl2C6H3 | CO2Me | 2.1g | 4.1gA (79) | anti-3.1gA (68)b |
| 12 | 3,4-Cl2C6H3 | CN | 2.2g | 4.2gA (6) | anti-3.2gA (63)b |
| 13 | 4-OMeC6H4 | CN | 2.2h | — | anti-3.2hA (67) |

To explain the remarkable stereoselectivity experienced in the formation of 1,3-thiazines during this study, a plausible mechanism is delineated in Scheme 3. It may be reasoned that the initial nucleophilic attack of sulfur results into a sp2-hybridized enolate intermediate. On the basis of literature reports,15 it is believed that the geometry of the intermediate enolate has the aryl group in the axial position due to the allylic strain. Subsequent protonation of the enolate from the less hindered side is presumed to be the kinetic step affording the kinetically-controlled syn-isomer. Due to the steric crowding, however, the syn-isomer is expected to be less stable and therefore transforms to a more stable anti-isomer. Alternatively, if the protonation of the enolate takes place from the more hindered side, the two bulky groups (electron withdrawing and aryl groups) prefer to go into diaxial position leading to thermodynamically stable anti-isomer. This implies that the one-pot formation of the anti-thiazines proceeds sequentially viathiourea formation, its transformation to syn-isomer and final stabilization to anti-product. However, we found it difficult to monitor the progress of reaction viaTLC since thioureas and corresponding thiazines have similar Rf values. In order to chemically assert our speculative pathway, therefore, a model study with 2.1aA was designed. We envisaged that a continuous monitoring of the reaction between 2.1a and AviaHPLC at different time-intervals may assist to delineate the sequence of events involved in the transformation of 2.1a to anti-3.1aA. Working toward this goal we considered essential to isolate the thiourea4.1aA in pure form. After brief optimization, we succeeded in preparing 4.1aA in pure form by carrying out the reaction between 2.1a and A at 0 °C for 3 h (Scheme 4).
 | ||
| Scheme 3 Plausible mechanism leading to the formation of anti 2-susbstituted amino-5,6-dihydro-4H-1,3-thiazines. | ||
 | ||
| Scheme 4 Synthesis of allyl thiourea from 2.1a. | ||
The HPLC (under isocratic condition) of 4.1aA and anti-3.1aA indicated the retention time of 3.5 and 4.8 min, respectively. Subsequently 2.1a was treated with A at room temperature and the reaction was monitored viaHPLC. The first aliquot from the reaction was drawn after 30 min and subjected to HPLC. Interestingly the HPLC chromatogram contained three peaks out of which two corresponded to 4.1aA and anti-3.1aA. The third peak at 4.0 min was expected to be for the syn-3.1aA. The HPLC analysis of the second aliquot drawn after 2 h revealed the presence of syn-3.1a and anti-3.1aA in approximately 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio which was corroborated by recording 1H NMR of the mixture (see supporting information†). Analysis of the third aliquot that was drawn at 6 h showed the presence of peak corresponding to the anti-3.1aA exclusively. This study clearly demonstrated that the thiourea initially produces the syn-product which transforms to more stable anti form during the reaction.
:1 ratio which was corroborated by recording 1H NMR of the mixture (see supporting information†). Analysis of the third aliquot that was drawn at 6 h showed the presence of peak corresponding to the anti-3.1aA exclusively. This study clearly demonstrated that the thiourea initially produces the syn-product which transforms to more stable anti form during the reaction.
In order to explain the formation of syn-1,3-thiazines from the allylamines of 2-halosubstituted benzaldehydes, we speculate that there is a chelation of In with enolate on one hand and halogen on the other resulting in a cyclic intermediate which restricts kinetic protonation leading to the syn-product exclusively (Scheme 5). However if the halo group is placed at 3 or/and 4-position of the phenyl ring it cannot participate in such interaction thereby affording only the anti-isomer which is the thermodynamic product.
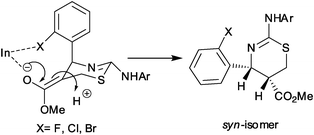 | ||
| Scheme 5 Stabilization of the enolate intermediate in the presence of InCl3 leading to kinetic controlled syn-isomer. | ||
In order to assess whether the trend displayed by different allylamines towards A extended to other aryl isothiocyanates (B,C), more reactions were performed. Accordingly amine 2.1b and 2.2b were treated with 2-bromophenyl isothiocyanate (C) whereas amine2.1f was treated with 4-chlorophenyl isothiocyanate (B). It was satisfying to find that similar to earlier observation, 2.1b and 2.2b react with C to afford the corresponding thiazinesanti-3.1bC and anti-3.2bCvia cascade sequence while 2.1f and 2.2f react with B to initially afford the thiourea4.1fB and 4.2fB which smoothly transforms to thiazineanti-3.1fB and anti-3.2fB, respectively viaInCl3-mediated intramolecular cyclization (Table 3). Additionally we found that arresting the reaction of 2.1b with C in 30 min affords a mixture of syn-3.1bC and anti-3.1bC in 0.3:1 ratio as analyzed by 1H NMR (see supporting information†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | Ar (no.) | EWG | 4 (yield %) | 3 (yield %)a |
| a Isolated yields after column chromatography. b Yields are from two-step one-pot reaction. | |||||
| 1 | 2.1b | 2-BrC6H4 (C) | CO2Me | — | anti-3.1bC (68) |
| 2 | 2.2b | 2-BrC6H4 (C) | CN | — | anti-3.2bC (71) |
| 3 | 2.1f | 4-ClC6H4 (B) | CO2Me | 4.1fB (89) | anti-3.1fB (72)b |
| 4 | 2.2f | 4-ClC6H4 (B) | CN | 4.2fB (80) | anti-3.2fB (68)b |

Conclusions
In summary, we have developed a simple and practical route for stereoselective synthesis of diverse 2-aryl amino 5,6-dihydro-4H-1,3-thiazines from the primary allylamines prepared from the MBH acetates. The reaction of aryl isothiocyanates with amines proceeds via initial thiourea formation followed by in situ sulfa-Michael reaction leading to intramolecular cyclization. It was observed that reaction is fast for amines derived from the MBH adduct of benzaldehyde or electron donating group containing benzaldehydes to afford anti-products exclusively in a cascade sequence. In contrast the reaction of amines derived from the MBH adducts of electron withdrawing group bearing benzaldehydes with aryl isothiocyanates initially afford thioureas, which require Lewis-acid to induce the sulfa-Michael reaction for cyclization to yield syn or anti-products based on the position of the substituent in the phenyl ring. Nevertheless the two-step sequence is completed as a one-pot procedure. In order to explain the remarkable stereoselectivity a plausible mechanism has been offered wherein it is suggested that the syn isomer is the kinetic product whereas the trans isomer is the thermodynamic product. A proof of concept for the mechanism by closely monitoring the progress of reaction with HPLC and trapping of the intermediates is provided. This work enhances the repertoire of the pharmacologically relevant heterocycles which can be synthesized from the MBH chemistry. Further application of sulfa-Michael reaction in the MBH derivatives to achieve the synthesis of diverse sulfur-heterocycles is currently underway in our laboratory.Experimental
General experimental
Melting points are uncorrected and were determined in capillary tubes on a Precision melting point apparatus containing silicon oil. IR spectra were recorded using a Perkin Elmer's RX I FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded either on a Bruker DPX-200 FT or Bruker Avance DRX-300 spectrometer, using TMS as an internal standard (chemical shifts in δ). The ESMS were recorded on MICROMASS Quadro-II LCMS system. The HRMS spectra were recorded as EI-HRMS on a JEOL system or as DART-HRMS (recorded as ES+) on a JEOL-AccuTOF JMS-T100 LC Mass spectrometer having DART (Direct Analysis in Real Time) source using 35Cl and 79Br for the mass calculation. Elemental analyses were performed on a Carlo Erba's 108 or an Elementar's Vario EL III microanalyzer. The HPLC were performed on Merck Hitachi D 7000 system having a Develosil C8 column with a particle size of 5 μ in isocratic solution of 5M aq. NH4OAc and MeCN. The flow rate was maintained at 0.5 mL/min for 20 min. The room temperature varied between 21 °C and 35 °C. All solvents and reagents were used as supplied by the vendors without any further purification. All column chromatography were performed on silica gel 100–200 mesh otherwise stated. The yields for allyl thioureas and 1.3-thiazines were calculated based on the respective MBH acetate as the starting substrate.Specific experimental
:1, 20 mL) was added DABCO (0.57 g, 5.12 mmol) and the reaction mixture was stirred for 10 min at room temperature (30 °C). After the reaction mixture became clear, aqueous ammonia (30%, 20 mL) was added at the same temperature, and the reaction was continued. After 30 min, the organic phase was separated and the aqueous phase was further extracted with CH2Cl2 (10 mL). The combined organic layer containing the allylamine2.1a was dried over anhydrous Na2SO4 and instantly used for further reaction.
:9) furnished pure product anti-3.1aA (0.61 g, 61%) as a white solid.
:20, v/v); νmax (KBr) 1716 (CO2Me), 3369 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.92–2.99 (m, 1H, CH), 3.09 (dd, 1H, J1 = 12.3 Hz, J2 = 3.8 Hz, CH2), 3.35 (dd, 1H, J1 = 13.5 Hz, J2 = 4.5 Hz, CH2), 3.57 (s, 3H, OCH3), 4.98 (d, 1H, J = 7.7 Hz, CH), 7.00 (s, 1H, NH), 7.25 (d, 5H, J = 6.0 Hz, ArH), 7.32–7.37 (m, 5H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.2, 50.5, 51.8, 52.3, 121.0, 123.1, 127.2, 127.5, 127.6, 127.7, 127.9, 128.5, 128.7, 129.0, 136.9, 142.1, 166.7, 172.4; mass (ES+) m/z = 327.1 (M++1). DART-HRMS Calcd. for C18H19N2O2S 327.1167; Found 327.1169.
:20, v/v); νmax (Neat) 1715 (CO2Me), 3370 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.93–2.99 (m, 1H, CH), 3.06 (dd, 1H, J1 = 12.5 Hz, J2 = 3.8 Hz, CH2), 3.32 (dd, 1H, J1 = 12.4 Hz, J2 = 8.5 Hz, CH2), 3.58 (s, 3H, OCH3), 4.95 (d, 1H, J = 7.4 Hz, CH), 7.09 (d, 2H, J = 8.6 Hz, ArH), 7.18 (d, 2H, J = 8.7 Hz, ArH), 7.30–7.35 (m, 6H, ArH and NH); 13C NMR (75 MHz, CDCl3) δ = 26.9, 45.1, 52.3, 59.5, 122.8, 127.0, 127.3, 127.7, 128.0, 128.5, 128.8, 128.9, 141.5, 142.8, 150.7, 171.9; mass (ES+) m/z = 361.0 (M++1), 363.1 (M++3). DART-HRMS Calcd. for C18H18ClN2O2S 361.0778; Found 361.0781.
:20, v/v); νmax (Neat) 1732 (CO2Me), 3372 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.01 (brs, 1H, CH), 3.09 (dd, 1H, J1 = 12.4 Hz, J2 = 3.7 Hz, CH2), 3.35 (dd, 1H, J1 = 12.7 Hz, J2 = 8.4 Hz, CH2), 3.60 (s, 3H, OCH3), 5.02 (d, 1H, J = 6.9 Hz, CH), 6.87 (t, 1H, J = 8.1 Hz, ArH), 7.18 (t, 2H, J = 7.8 Hz, ArH), 7.32–7.38 (m, 5H, ArH), 7.52 (d, 1H, J = 8.0 Hz, ArH), 7.62 (brs, 1H, NH); 13C NMR (75 MHz, CDCl3) δ = 26.9, 51.8, 52.4, 59.4, 124.3, 126.7, 127.0, 127.3, 127.5, 127.7, 128.1, 128.7, 128.9, 129.8, 132.7, 137.0, 137.5, 171.8; mass (ES+) m/z = 405.0 (M++1), 407.0 (M++3). DART-HRMS Calcd. for C18H18BrN2O2S 405.0272; Found 405.0273.
:20, v/v); νmax (KBr) 2210 (CN), 3337 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.05 (t, 1H, J = 6.5 Hz, CH), 3.20–3.26 (m, 2H, CH2), 5.00 (d, 1H, J = 6.8 Hz, CH), 7.03 (t, 1H, J = 7.3 Hz,NH), 7.26 (t, 3H, J = 5.8 Hz, ArH), 7.34–7.44 (m, 7H, ArH); 13C NMR (50 MHz, CDCl3) δ = 26.9, 32.0, 60.6, 120.8, 123.6, 125.3, 127.2, 128.5, 129.0, 129.1, 140.6,145.0; mass (ES+) m/z = 294.1 (M++1). DART-HRMS Calcd. for C17H16N3S 294.1065; Found 294.1066.
:20, v/v); νmax (Neat) 2209 (CN), 3368 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.07 (d, 1H, J = 4.5 Hz, CH), 3.20–3.30 (m, 2H, CH2), 4.97 (d, 1H, J = 6.5 Hz, CH), 7.18–7.26 (m, 4H, ArH), 7.35–7.42 (m, 6H, ArH); 13C NMR (75 MHz, CDCl3) δ = 27.0, 45.9, 52.0, 121.4, 121.8, 123.3, 123.6, 127.1, 127.3, 127.9, 128.1, 128.5, 128.7, 128.9, 129.0, 141.7, 172.1; mass (ES+) m/z = 328.0 (M++1), 330.0 (M++3). DART-HRMS Calcd. for C17H15ClN3S 328.0675; Found 328.0679.
:20, v/v); νmax (Neat) 2214 (CN), 3369 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.10–3.12 (m, 1H, CH), 3.20–3.28 (m, 2H, CH2), 5.01 (d, 1H, J = 6.8 Hz, CH), 6.87–6.92 (m, 1H, ArH), 7.19 (t, 1H, J = 8.1 Hz, ArH), 7.36–7.41 (m, 6H, ArH), 7.53 (d, 1H, J = 8.0 Hz, ArH), 7.92 (brs, 1H, NH); 13C NMR (75 MHz, CDCl3) δ = 26.9, 32.2, 60.3, 118.7, 122.6, 123.8, 124.4, 127.2, 127.4, 128.3, 128.7, 129.2, 132.6, 140.1, 148.6; mass (ES+) m/z = 372.0 (M++1), 374.0 (M++3). DART-HRMS Calcd. for C17H15BrN3S 372.0170; Found 372.0173.
:20, v/v); νmax (Neat) 1712 (CO2Me), 3419 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.35 (s, 3H, CH3), 2.84–2.96 (m. 1H, CH), 3.08 (dd, 1H, J1 = 12.5 Hz, J2 = 3.8 Hz, CH2), 3.35 (1H, J1 = 12.5 Hz, J2 = 8.8 Hz, CH2), 3.58 (s, 3H, OCH3), 4.94 (d, 1H, J = 7.9 Hz, CH), 7.00 (brs, 1H, NH), 7.13–7.18 (m, 3H, ArH), 7.23–7.26 (m, 5H, ArH), 7.33 (d, 1H, J = 7.4 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 21.3, 27.3, 52.1, 52.3, 59.7, 121.3, 123.2, 123.3, 127.0, 127.3,129.0, 129.2, 129.5, 136.4, 137.7, 138.7, 149.6, 172.2; mass (ES+) m/z = 341.1 (M++1). DART-HRMS calcd. for C19H21N2O2S 341.1324; Found 341.1329.
:20, v/v); νmax (KBr) 2219 (CN), 3372 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.43 (s, 3H, CH3), 3.19–3.25 (m. 1H, CH), 3.27–3.37 (m, 2H, CH2), 5.05 (d, 1H, J = 6.8 Hz, CH), 7.33–7.41 (m, 7H, ArH), 7.49–7.54 (m, 2H, ArH), 8.40 (brs, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 21.2, 26.6, 32.1, 60.6, 118.5, 121.9, 125.1, 126.9, 129.2, 129.8, 130.0, 130.2, 133.8, 136.1, 138.8, 150.9; mass (ES+) m/z = 308.1 (M++1). DART-HRMS calcd. for C18H18N3S 308.1221; Found 308.1229.
:20, v/v); νmax (KBr) 1723 (CO2Me), 3406 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.33 (s, 3H, CH3), 2.99–3.08 (m. 2H, CH), 3.30 (dd, 1H, J1 = 12.7 Hz, J2 = 8.2 Hz, CH2), 3.59 (s, 3H, OCH3), 4.94 (d, 1H, J = 6.7 Hz, CH), 6.82–6.88 (m,1H, ArH), 7.13–7.18 (m, 3H, ArH), 7.22–7.25 (m, 2H, ArH), 7.45–7.51 (m, 2H, ArH); 13C NMR (50 MHz, CDCl3) δ = 21.2, 23.0, 27.1, 52.1, 52.4, 123.3, 124.1, 126.9, 127.2, 128.1, 129.3, 129.5, 132.6, 136.1, 137.9, 171.8; mass (ES+) m/z = 419.1 (M++1), 421.2 (M++3). DART-HRMS calcd. for C19H20BrN2O2S 419.0429; Found 419.0419.
:20, v/v); νmax (KBr) 2209 (CN), 3379 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.37 (s, 3H, CH3), 3.05–3.12 (m. 1H, CH), 3.23–3.26 (m, 2H, CH2), 4.95 (d, 1H, J = 7.1 Hz, CH), 6.85–6.91 (m, 1H, ArH), 7.16–7.29 (m, 6H, ArH), 7.51–7.54 (m, 1H, ArH), 7.85 (s, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 21.2, 27.0, 55.5, 59.9, 114.4, 118.9, 121.1, 122.7, 123.8, 124.3, 127.0, 128.3, 129.1, 129.8, 132.3, 159.8; mass (ES+) m/z = 386.1 (M++1), 388.2 (M++3). DART-HRMS calcd. for C18H17BrN3S 386.0387; Found 386.0388.
:20, v/v); νmax (KBr) 2225 (CN),3331 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.90–2.97 (m, 1H, CH), 3.21–3.37 (m, 2H, CH2), 4.94 (d, 1H, J = 7.8 Hz, CH), 7.03 (t, 1H, J = 7.5 Hz, NH), 7.26 (t, 3H, J = 9.6 Hz, ArH), 7.33–7.41 (m, 6H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.2, 32.1, 60.4, 118.8, 120.6, 123.7, 128.7, 129.1, 134.3, 139.4, 140.1, 151.9; mass (ES+) m/z = 328.0 (M++1), 330.0 (M++3). DART-HRMS calcd. for C17H15ClN3S 328.0675; Found 328.0673.
:20, v/v); νmax (KBr) 2216(CN), 3395 (NH) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.96–3.06 (m. 1H, CH), 3.23–3.27 (m, 2H, CH2), 3.83 (s, 3H, OCH3), 4.92 (d, 1H, J = 7.2 Hz, CH), 6.91–7.07 (m, 3H, ArH), 7.21–7.33 (m, 7H, ArH and NH); 13C NMR (50 MHz, CDCl3) δ = 27.0, 32.4, 55.5, 60.0, 114.4, 119.0, 120.9, 123.5, 128.4, 129.1, 132.6, 159.8; mass (ES+) m/z = 324.1 (M++1). DART-HRMS calcd. for C18H18N3OS 324.1171; Found 324.1170.
:30, v/v); νmax (KBr) 1638 (CS), 1730 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.67 (s, 3H, OCH3), 5.98 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.40 (s, 1H, CH2), 6.80 (d, 1H, J = 7.5 Hz, CH), 7.10–7.16 (m, 1H, NH), 7.25–7.33 (m, 6H, ArH), 7.44 (t, 2H, J = 7.4 Hz, ArH), 7.57 (d, 1H, J = 7.8 Hz, ArH), 7.90 (s, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 52.2, 60.0, 124.0, 125.1, 125.2, 127.4, 127.7, 128.8, 129.3, 129.5, 130.2, 133.6, 136.2, 137.1, 137.9, 166.4, 180.2; mass (ES+) m/z = 405.0 (M++1), 407.0 (M++3). Anal. Calcd. for C18H17BrN2O2S (Exact mass: 404.0194); C, 53.34; H, 4.23; N, 6.91; Found C, 53.13; H, 4.39; N, 6.62.
:9) furnished the pure products as oils or solids.
:9) furnished pure product syn-3.1cA (0.50 g, 77%) as a white solid.
:20, v/v); νmax (KBr) 1717 (CO2Me), 3371 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.03 (dd, 1H, J1 = 12.5 Hz, J2 = 3.2 Hz, CH2), 3.13 (brs, 1H, CH), 3.26 (dd, 1H, J1 = 12.4 Hz, J2 = 5.9 Hz, CH2), 3.73 (s, 3H, OCH3), 5.65 (d, 1H, J = 2.2 Hz, CH), 6.98 (t, 1H, J = 6.8 Hz, ArH), 7.00–7.24 (m, 5H, ArH), 7.32–7.42 (m, 2H, ArH), 7.55 (d, 1H, J = 7.8 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 22.7, 41.3, 52.6, 57.8, 121.5, 122.5, 123.3, 124.9, 127.2, 127.8, 128.9, 129.4, 129.9, 133.2, 140.8, 142.4, 150.2, 171.2; mass (ES+) m/z = 405.0 (M++1), 407.0 (M++3). DART-HRMS calcd. for C18H17BrN2O2S 405.0272; Found 405.0277.
:20, v/v); νmax (KBr) 2215 (CN), 3317 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.13 (d, 2H, J = 4.6 Hz, CH2), 3.32–3.36 (m, 1H, CH), 5.58 (d, 1H, J = 4.0 Hz, CH), 7.03 (t, 1H, J = 7.2 Hz, CH), 7.19–7.42 (m, 8H, ArH), 7.61 (d, 1H, J = 7.8 Hz, NH); 13C NMR (50 MHz, CDCl3) δ = 25.1, 27.6, 59.3, 118.7, 120.9, 122.7, 123.6, 128.0, 129.0, 129.6, 129.9, 133.6, 139.4, 140.2, 147.6; mass (ES+) m/z = 372.0 (M++1), 374.0 (M++3). DART-HRMS Calcd. for C17H15BrN3S 372.0170; Found 372.0171.
:20, v/v); νmax (KBr) 1731 (CO2Me), 3369 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.02–3.14 (m, 2H, CH and CH2), 3.26–3.30 (m, 1H, CH2), 3.73 (s, 3H, OCH3), 5.70 (d, 1H, J = 3.8 Hz, CH), 7.00 (t, 1H, J = 7.1 Hz, ArH), 7.21–7.42 (m, 8H, ArH); 13C NMR (75 MHz, CDCl3) δ = 25.2, 41.4, 52.6, 56.1, 120.9, 122.5, 123.1, 123.6, 127.2, 129.0, 129.4, 129.9, 131.7, 132.4, 134.0, 139.7, 148.9, 171.5; mass (ES+) m/z = 361.1 (M++1), 363.2 (M++3). DART-HRMS Calcd. for C18H18ClN2O2S 361.0778; Found 361.0783.
:20, v/v); νmax (KBr) 2226 (CN), 3367 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.37 (d, 1H, J = 10.0 Hz, CH), 3.67–3.73 (m, 2H, CH2), 5.13 (d, 1H, J = 3.2 Hz, CH), 7.04 (t, 1H, J = 8.4 Hz, ArH), 7.26–7.33 (m, 4H, ArH), 7.39–7.45 (m, 2H, ArH), 7.52 (d, 2H, J = 7.9 Hz, ArH), 7.84 (d, 1H, J = 7.9 Hz, NH); 13C NMR (50 MHz, CDCl3) δ = 28.4, 29.7, 57.2, 117.2, 120.4, 123.4, 127.8, 129.1, 129.3, 129.4, 130.1, 132.0, 138.0, 139.9, 148.1; mass (ES+) m/z = 328.1 (M++1), 330.2 (M++3). DART-HRMS Calcd. for C17H15ClN3S 328.0675; Found 328.0680.
:20, v/v); νmax (KBr) 1738 (CO2Me), 3412 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08 (dd, 1H, J1 = 12.3 Hz, J2 = 2.5 Hz, CH2), 3.31–3.39 (m, 2H, CH and CH2), 3.83 (s, 3H, OCH3), 5.66 (d, 1H, J = 3.8 Hz, CH), 6.14 (t, 1H, J = 10.0 Hz, ArH), 7.24–7.29 (m, 1H, ArH), 7.33–7.44 (m, 7H, ArH), 11.93 (brs, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 24.6, 41.5, 51.0, 53.3, 116.3 (J = 20.9 Hz), 125.0 (J = 19.3 Hz), 126.3, 128.1, 128.8, 129.7, 131.1 (J = 8.2 Hz), 132.9, 134.3, 142.8, 161.8, 166.6, 168.9; mass (ES+) m/z = 345.0 (M++1). DART-HRMS calcd. for C18H18FN2O2S 345.1073; Found 345.1067.
:20, v/v); νmax (KBr) 2221 (CN), 3347 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.35 (dd, 1H, J1 = 18.5 Hz, J2 = 5.2 Hz, CH2), 3.51–3.57 (m, 1H, CH), 3.66 (dd, 1H, J1 = 18.5 Hz, J2 = 6.8 Hz, CH2), 5.14 (d, 1H, J = 4.8 Hz, CH), 7.01–7.20 (m, 2H, ArH), 7.23–7.41 (m, 5H, ArH), 7.50–7.54 (m, 2H, ArH), 7.73–7.82 (m, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 26.1, 29.1, 54.1 (J = 42.2 Hz), 115.7, 118.6, 120.6 (J = 18.6 Hz), 123.5 (J = 9.5 Hz), 124.7 (J = 3.4 Hz), 124.9 (J = 3.1 Hz), 127.8, 129.0, 129.2 (J = 3.6 Hz), 129.6, 130.1 (J = 8.3 Hz), 157.4, 167.0; mass (ES+) m/z = 312.1 (M++1). DART-HRMS calcd. for C17H15FN3S 312.0971; Found 312.0966.
:20, v/v); νmax (KBr) 1710 (CO2Me), 3319 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.78–2.82 (m, 1H, CH), 3.05 (dd, 1H, J1 = 12.5 Hz, J2 = 3.9 Hz, CH2), 3.36 (dd, 1H, J1 = 12.3 Hz, J2 = 9.3 Hz, CH2), 3.56 (s, 3H, OCH3), 4.89 (d, 1H, J = 8.0 Hz, CH), 7.00 (brs, 1H, ArH), 7.16 (d, 1H, J = 8.3 Hz, ArH), 7.23–7.32 (m, 8H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.3, 45.9, 52.1, 52.3, 121.0, 121.4, 123.2, 123.3, 128.5, 128.6, 128.8, 128.9, 133.5, 133.7, 140.7, 149.7, 172.3; mass (ES+) m/z = 361.0 (M++1), 363.1 (M++3). DART-HRMS Calcd. for C18H18ClN2O2S 361.0778; Found 361.0781.
:20, v/v); νmax (KBr) 1729 (CO2Me), 3362 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.77 (brs, 1H, CH), 3.08 (dd, 1H, J1 = 12.5 Hz, J2 = 4.1 Hz, CH2), 3.40 (dd, 1H, J1 = 12.4 Hz, J2 = 9.5 Hz, CH2), 3.59 (s, 3H, OCH3), 4.91 (d, 1H, J = 8.1 Hz, CH), 7.00–7.03 (m, 1H, NH), 7.16–7.33 (m, 6H, ArH), 7.42 (d, 2H, J = 9.0 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.3, 45.6, 52.4, 59.8, 120.8, 123.3, 126.7, 128.9, 129.4, 129.6, 130.5, 131.6, 132.6, 141.8, 142.8, 149.0, 172.3; mass (ES+) m/z = 395.0 (M++1), 397.1 (M++3). DART-HRMS calcd. for C18H17Cl2N2O2S 395.0388; Found 395.0393.
:20, v/v); νmax (KBr) 2218 (CN), 3365 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.88–2.95 (m, 1H, CH), 3.25 (dd, 1H, J1 = 12.9 Hz, J2 = 4.1 Hz, CH2), 3.35 (dd, 1H, J1 = 12.7 Hz, J2 = 8.9 Hz, CH2), 4.90 (d, 1H, J = 8.0 Hz, CH), 7.05 (t, 1H, J = 7.4 Hz, ArH), 7.26 (t, 5H, J = 9.5 Hz, ArH), 7.39 (d, 1H, J = 7.8 Hz, ArH), 7.49 (d, 1H, J = 9.6 Hz, ArH); 13C NMR (50 MHz, DMSO-d6) δ = 25.3, 28.8, 58.2, 118.9, 119.8, 121.6, 127.7, 128.5, 129.5, 130.2, 130.6, 131.0, 141.0, 142.9, 144.9, 146.6; mass (ES+) m/z = 362.0 (M++1), 364.1 (M++3). DART-HRMS calcd. for C17H14Cl2N3S 362.0285; Found 362.0279.
:20, v/v); νmax (Neat) 1728 (CO2Me), 3386 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.81–2.91 (m, 1H, CH), 3.00–3.09 (m, 1H, CH2), 3.28–3.39 (m, 1H, CH2), 3.58 (s, 3H, OCH3), 4.88 (d, 1H, J = 7.7 Hz, CH), 7.07–7.33 (m, 9H, ArH and NH); 13C NMR (50 MHz, CDCl3) δ = 27.2, 45.9, 52.5, 59.4, 122.7, 122.9, 128.5, 128.8, 129.0, 133.9, 140.1, 141.6, 150.7, 171.9; mass (ES+) m/z = 395.0 (M++1), 397.1 (M++3). DART-HRMS Calcd. for C18H17Cl2N2O2S 395.0388; Found 395.0396.
:20, v/v); νmax (KBr) 2219 (CN), 3374 (NH) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.99–3.08 (m, 1H, CH), 318–3.38 (m, 2H, CH2), 4.96 (d, 1H, J = 7.3 Hz, CH), 7.00 (s, 1H, NH), 7.21–7.26 (m, 2H, ArH and NH), 7.29–7.43 (m, 7H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.0, 29.9, 59.0, 118.6, 122.3, 128.7, 128.8, 129.2, 129.23, 123.26, 129.4, 134.6, 134.7, 138.2, 138.8; mass (ES+) m/z = 363.0 (M++1), 365.1 (M++3). DART-HRMS Calcd. for C17H14Cl2N3S 363.2842; Found 363.2840.
CH2), 6.40 (s, 1H, CH2), 6.80 (d, 1H, J = 7.5 Hz, CH), 7.10–7.16 (m, 1H, NH), 7.25–7.33 (m, 6H, ArH), 7.44 (t, 2H, J = 7.4 Hz, ArH), 7.57 (d, 1H, J = 7.8 Hz, ArH), 7.90 (s, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 52.2, 60.0, 124.0, 125.1, 125.2, 127.4, 127.7, 128.8, 129.3, 129.5, 130.2, 133.6, 136.2, 137.1, 137.9, 166.4, 180.2; mass (ES+) m/z = 405.0 (M++1), 407.0 (M++3). Anal. Calcd. for C18H17BrN2O2S (Exact mass: 404.0194); C, 53.34; H, 4.23; N, 6.91; Found C, 53.13; H, 4.39; N, 6.62.
:9) furnished the pure products as oils or solids.
:9) furnished pure product syn-3.1cA (0.50 g, 77%) as a white solid.
:20, v/v); νmax (KBr) 1717 (CO2Me), 3371 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.03 (dd, 1H, J1 = 12.5 Hz, J2 = 3.2 Hz, CH2), 3.13 (brs, 1H, CH), 3.26 (dd, 1H, J1 = 12.4 Hz, J2 = 5.9 Hz, CH2), 3.73 (s, 3H, OCH3), 5.65 (d, 1H, J = 2.2 Hz, CH), 6.98 (t, 1H, J = 6.8 Hz, ArH), 7.00–7.24 (m, 5H, ArH), 7.32–7.42 (m, 2H, ArH), 7.55 (d, 1H, J = 7.8 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 22.7, 41.3, 52.6, 57.8, 121.5, 122.5, 123.3, 124.9, 127.2, 127.8, 128.9, 129.4, 129.9, 133.2, 140.8, 142.4, 150.2, 171.2; mass (ES+) m/z = 405.0 (M++1), 407.0 (M++3). DART-HRMS calcd. for C18H17BrN2O2S 405.0272; Found 405.0277.
:20, v/v); νmax (KBr) 2215 (CN), 3317 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.13 (d, 2H, J = 4.6 Hz, CH2), 3.32–3.36 (m, 1H, CH), 5.58 (d, 1H, J = 4.0 Hz, CH), 7.03 (t, 1H, J = 7.2 Hz, CH), 7.19–7.42 (m, 8H, ArH), 7.61 (d, 1H, J = 7.8 Hz, NH); 13C NMR (50 MHz, CDCl3) δ = 25.1, 27.6, 59.3, 118.7, 120.9, 122.7, 123.6, 128.0, 129.0, 129.6, 129.9, 133.6, 139.4, 140.2, 147.6; mass (ES+) m/z = 372.0 (M++1), 374.0 (M++3). DART-HRMS Calcd. for C17H15BrN3S 372.0170; Found 372.0171.
:20, v/v); νmax (KBr) 1731 (CO2Me), 3369 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.02–3.14 (m, 2H, CH and CH2), 3.26–3.30 (m, 1H, CH2), 3.73 (s, 3H, OCH3), 5.70 (d, 1H, J = 3.8 Hz, CH), 7.00 (t, 1H, J = 7.1 Hz, ArH), 7.21–7.42 (m, 8H, ArH); 13C NMR (75 MHz, CDCl3) δ = 25.2, 41.4, 52.6, 56.1, 120.9, 122.5, 123.1, 123.6, 127.2, 129.0, 129.4, 129.9, 131.7, 132.4, 134.0, 139.7, 148.9, 171.5; mass (ES+) m/z = 361.1 (M++1), 363.2 (M++3). DART-HRMS Calcd. for C18H18ClN2O2S 361.0778; Found 361.0783.
:20, v/v); νmax (KBr) 2226 (CN), 3367 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.37 (d, 1H, J = 10.0 Hz, CH), 3.67–3.73 (m, 2H, CH2), 5.13 (d, 1H, J = 3.2 Hz, CH), 7.04 (t, 1H, J = 8.4 Hz, ArH), 7.26–7.33 (m, 4H, ArH), 7.39–7.45 (m, 2H, ArH), 7.52 (d, 2H, J = 7.9 Hz, ArH), 7.84 (d, 1H, J = 7.9 Hz, NH); 13C NMR (50 MHz, CDCl3) δ = 28.4, 29.7, 57.2, 117.2, 120.4, 123.4, 127.8, 129.1, 129.3, 129.4, 130.1, 132.0, 138.0, 139.9, 148.1; mass (ES+) m/z = 328.1 (M++1), 330.2 (M++3). DART-HRMS Calcd. for C17H15ClN3S 328.0675; Found 328.0680.
:20, v/v); νmax (KBr) 1738 (CO2Me), 3412 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.08 (dd, 1H, J1 = 12.3 Hz, J2 = 2.5 Hz, CH2), 3.31–3.39 (m, 2H, CH and CH2), 3.83 (s, 3H, OCH3), 5.66 (d, 1H, J = 3.8 Hz, CH), 6.14 (t, 1H, J = 10.0 Hz, ArH), 7.24–7.29 (m, 1H, ArH), 7.33–7.44 (m, 7H, ArH), 11.93 (brs, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 24.6, 41.5, 51.0, 53.3, 116.3 (J = 20.9 Hz), 125.0 (J = 19.3 Hz), 126.3, 128.1, 128.8, 129.7, 131.1 (J = 8.2 Hz), 132.9, 134.3, 142.8, 161.8, 166.6, 168.9; mass (ES+) m/z = 345.0 (M++1). DART-HRMS calcd. for C18H18FN2O2S 345.1073; Found 345.1067.
:20, v/v); νmax (KBr) 2221 (CN), 3347 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 3.35 (dd, 1H, J1 = 18.5 Hz, J2 = 5.2 Hz, CH2), 3.51–3.57 (m, 1H, CH), 3.66 (dd, 1H, J1 = 18.5 Hz, J2 = 6.8 Hz, CH2), 5.14 (d, 1H, J = 4.8 Hz, CH), 7.01–7.20 (m, 2H, ArH), 7.23–7.41 (m, 5H, ArH), 7.50–7.54 (m, 2H, ArH), 7.73–7.82 (m, 1H, NH); 13C NMR (50 MHz, CDCl3) δ = 26.1, 29.1, 54.1 (J = 42.2 Hz), 115.7, 118.6, 120.6 (J = 18.6 Hz), 123.5 (J = 9.5 Hz), 124.7 (J = 3.4 Hz), 124.9 (J = 3.1 Hz), 127.8, 129.0, 129.2 (J = 3.6 Hz), 129.6, 130.1 (J = 8.3 Hz), 157.4, 167.0; mass (ES+) m/z = 312.1 (M++1). DART-HRMS calcd. for C17H15FN3S 312.0971; Found 312.0966.
:20, v/v); νmax (KBr) 1710 (CO2Me), 3319 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.78–2.82 (m, 1H, CH), 3.05 (dd, 1H, J1 = 12.5 Hz, J2 = 3.9 Hz, CH2), 3.36 (dd, 1H, J1 = 12.3 Hz, J2 = 9.3 Hz, CH2), 3.56 (s, 3H, OCH3), 4.89 (d, 1H, J = 8.0 Hz, CH), 7.00 (brs, 1H, ArH), 7.16 (d, 1H, J = 8.3 Hz, ArH), 7.23–7.32 (m, 8H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.3, 45.9, 52.1, 52.3, 121.0, 121.4, 123.2, 123.3, 128.5, 128.6, 128.8, 128.9, 133.5, 133.7, 140.7, 149.7, 172.3; mass (ES+) m/z = 361.0 (M++1), 363.1 (M++3). DART-HRMS Calcd. for C18H18ClN2O2S 361.0778; Found 361.0781.
:20, v/v); νmax (KBr) 1729 (CO2Me), 3362 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.77 (brs, 1H, CH), 3.08 (dd, 1H, J1 = 12.5 Hz, J2 = 4.1 Hz, CH2), 3.40 (dd, 1H, J1 = 12.4 Hz, J2 = 9.5 Hz, CH2), 3.59 (s, 3H, OCH3), 4.91 (d, 1H, J = 8.1 Hz, CH), 7.00–7.03 (m, 1H, NH), 7.16–7.33 (m, 6H, ArH), 7.42 (d, 2H, J = 9.0 Hz, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.3, 45.6, 52.4, 59.8, 120.8, 123.3, 126.7, 128.9, 129.4, 129.6, 130.5, 131.6, 132.6, 141.8, 142.8, 149.0, 172.3; mass (ES+) m/z = 395.0 (M++1), 397.1 (M++3). DART-HRMS calcd. for C18H17Cl2N2O2S 395.0388; Found 395.0393.
:20, v/v); νmax (KBr) 2218 (CN), 3365 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.88–2.95 (m, 1H, CH), 3.25 (dd, 1H, J1 = 12.9 Hz, J2 = 4.1 Hz, CH2), 3.35 (dd, 1H, J1 = 12.7 Hz, J2 = 8.9 Hz, CH2), 4.90 (d, 1H, J = 8.0 Hz, CH), 7.05 (t, 1H, J = 7.4 Hz, ArH), 7.26 (t, 5H, J = 9.5 Hz, ArH), 7.39 (d, 1H, J = 7.8 Hz, ArH), 7.49 (d, 1H, J = 9.6 Hz, ArH); 13C NMR (50 MHz, DMSO-d6) δ = 25.3, 28.8, 58.2, 118.9, 119.8, 121.6, 127.7, 128.5, 129.5, 130.2, 130.6, 131.0, 141.0, 142.9, 144.9, 146.6; mass (ES+) m/z = 362.0 (M++1), 364.1 (M++3). DART-HRMS calcd. for C17H14Cl2N3S 362.0285; Found 362.0279.
:20, v/v); νmax (Neat) 1728 (CO2Me), 3386 (NH) cm−1; 1H NMR (300 MHz, CDCl3) δ = 2.81–2.91 (m, 1H, CH), 3.00–3.09 (m, 1H, CH2), 3.28–3.39 (m, 1H, CH2), 3.58 (s, 3H, OCH3), 4.88 (d, 1H, J = 7.7 Hz, CH), 7.07–7.33 (m, 9H, ArH and NH); 13C NMR (50 MHz, CDCl3) δ = 27.2, 45.9, 52.5, 59.4, 122.7, 122.9, 128.5, 128.8, 129.0, 133.9, 140.1, 141.6, 150.7, 171.9; mass (ES+) m/z = 395.0 (M++1), 397.1 (M++3). DART-HRMS Calcd. for C18H17Cl2N2O2S 395.0388; Found 395.0396.
:20, v/v); νmax (KBr) 2219 (CN), 3374 (NH) cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.99–3.08 (m, 1H, CH), 318–3.38 (m, 2H, CH2), 4.96 (d, 1H, J = 7.3 Hz, CH), 7.00 (s, 1H, NH), 7.21–7.26 (m, 2H, ArH and NH), 7.29–7.43 (m, 7H, ArH); 13C NMR (50 MHz, CDCl3) δ = 27.0, 29.9, 59.0, 118.6, 122.3, 128.7, 128.8, 129.2, 129.23, 123.26, 129.4, 134.6, 134.7, 138.2, 138.8; mass (ES+) m/z = 363.0 (M++1), 365.1 (M++3). DART-HRMS Calcd. for C17H14Cl2N3S 363.2842; Found 363.2840.
Acknowledgements
Two of the authors (SB and AM) gratefully acknowledge the financial support from Council of Scientific and Industrial Research, New Delhi in the form of fellowships. Authors gratefully acknowledge the SAIF Division of CDRI for recording all the spectroscopic and analytical data. This work was supported by a grant from Department of Science and Technology, New Delhi. Authors also acknowledge the comments from the reviewers which assisted to improve the manuscript considerably.References
- (a) J. M. Hamilton-Miller and T. W. Brumfitt, Infection, 1975, 3, 183–188 CrossRef CAS; (b) A. K. Miller, B. A. E. Celozzi, B. A. Pelak, E. O. Stapley and D. Hendlin, Antimicrob. Agents Chemother., 1972, 281–286 CAS.
- (a) M. Koketsu, T. Senda and H. Ishihara, Jpn Patent, 2000, 119263 ( Chem. Abstr. , 2000 , 132 , 293768 ) Search PubMed; (b) H. Kai, Y. Morioka, Y. Koriyama, K. Okamoto, Y. Hasegawa, M. Hattori, K. Koike, H. Chiba, S. Shinohara, Y. Iwamoto, K. Takahashi and N. Tanimoto, Bioorg. Med. Chem. Lett., 2008, 18, 6444–6447 CrossRef CAS.
- N. Karodia, In Comprehensive Heterocyclic Chemistry Vol. 4 (4.01 ed. A. R. Katrizky, C. A. Ramsden, R. J. K. Taylor, E. F. V. Scriven) Pergamon: London, 2008, 568–604. Search PubMed.
- A. Marchand, D. Mauger, A. Guingant and J. P. Pradere, Tetrahedron, 2004, 60, 1827–1839 CrossRef.
- A. Marchand, S. Collet, A. Guingant, J. P. Pradere and L. Toupet, Tetrahedron: Asymmetry, 1995, 6, 853–856 CrossRef CAS.
- N. Leflemme, P. Dallemagne and S. Rault, Tetrahedron Lett., 2004, 45, 1503–1505 CrossRef CAS.
- (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS , and references cited therein; (b) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS; (c) S. Gowrisankar, H. S. Lee, S. H. Kim, K. Y. Lee and J. N. Kim, Tetrahedron, 2009, 65, 8769–8780 CrossRef CAS; (d) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511–4574 CrossRef CAS.
- M. M. Sa, L. Fernandes, M. Ferreira and A. J. Bortoluzzi, Tetrahedron Lett., 2008, 49, 1228–1232 CrossRef CAS.
- L. D. S. Yadav, R. Patel and V. P. Srivastava, Tetrahedron Lett., 2009, 50, 1335–1339 CrossRef CAS.
- S. Nag, S. Bhowmik, H. Gauniyal and S. Batra, Eur. J. Org. Chem., 2010, 4705–4712 CrossRef CAS.
- (a) For Lewis acid mediated see S. Bonollo, D. Lanari, F. Pizzo and L. Vaccaro, Org. Lett., 2011, 2150–2152 CrossRef CAS; (b) C.-M. Chu, W.-J. Huang, C. Lu, P. Wu, J.-T. Liu and C.-F. Yao, Tetrahedron Lett., 2006, 47, 7375–7380 CrossRef CAS; (c) S. Gao, T. Tzeng, M. N. V. Sastry, C.-M. Chu, J.-T. Liu, C. Lin and C.-F Yao, Tetrahedron Lett., 2006, 47, 1889–1893 CrossRef CAS; (d) M. Dzurilla, P. Kutschy and P. Kristian, Synthesis, 1985, 933–934 CrossRef CAS; (e) N. K. Rana, S. Selvakumar, V. K. Singh and J. Org, Chem., 2010, 75, 2089–2091 CAS; (f) Y. Hui, J. Jiang, W. Wang, W. Chen, Y. Cai, L. Lin, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 4290–4293 CrossRef CAS; (g) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807–1821 RSC; (h) D. Enders, K. Luttgen and A. A. Narine, Synthesis, 2007, 959–980 CrossRef CAS; (i) P. Kotrusz and S. Toma, Molecules, 2006, 11, 197–205 CrossRef CAS; (j) A. P. Kozikowski and B. B. Mugrage, J. Org. Chem., 1989, 54, 2274–2275 CrossRef CAS.
- (a) A. Mishra, S. Hutait, S. Bhowmik, N. Rastogi, R. Roy and S. Batra, Synthesis, 2010, 2731–2748 CAS; (b) A. Mishra and S. Batra, Synthesis, 2009, 3077–3088 CAS; (c) R. Pathak, S. Nag and S. Batra, Synthesis, 2006, 4205–4211 CAS.
- Z. Chen and D. L. J. Clive, J. Org. Chem., 2010, 75, 7014–7017 CrossRef CAS.
- L. D. S. Yadav and A. Singh, Tetrahedron Lett., 2003, 44, 5637–5640 CrossRef CAS.
- (a) K. Chattopadhyay, R. Jana, V. W. Day, J. T. Douglas and J. A. Tunge, Org. Lett., 2010, 12, 3042–3045 CrossRef CAS; (b) H. E. Zimmerman, Acc. Chem. Res., 1987, 20, 263–265 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Spectral data for remaining compounds and copies of 1H- and 13C-NMR spectra for all compounds are included. See DOI: 10.1039/c1ra00399b/ |
| ‡ CDRI Communication no. 8106. |
| § Authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2011 |