Enable PVC plastic for a novel role: its functionalisation with diiron models of the sub-unit of [FeFe]–hydrogenase, assembly of film electrodes, and electrochemical investigations†
Lianjie
Wang
a,
Zhiyin
Xiao
b,
Xiang
Ru
a and
Xiaoming
Liu
*ab
aInstitute for Advanced Study/Department of Chemistry, Nanchang University, Nanchang, 330031, China. E-mail: xiaoming.liu@ncu.edu.cn; Fax: +86 (0)791 3969254; Tel: +86 (0)791 3969254
bCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, 314001, China. E-mail: xiaoming.liu@mail.zjxu.edu.cn; Fax: +86 (0)573 8364 3937; Tel: +86 (0)573 8364 3937
First published on 16th September 2011
Abstract
Three polymers functionalised with diiron carbonyl units, PVC–Fe-A, -B, and -C, were prepared using commercially available polymer PVC (polyvinyl chloride). PVC–Fe-A resulted from the reaction of the reduced form of a diiron complex, [Fe2(μ-S)2(CO)6], with PVC, whereas PVC–Fe-B and PVC–Fe-C were, respectively, prepared by reacting PVC–N3, the polymer functionalised with azide groups by substitution of the chloride of the polymer, with two diiron complexes, [Fe2(μ-SCH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2(CO)6] (1) and [Fe2(μ-SnBut)(μ-SCH2CCH)(CO)6] (2, nBut = –CH2CH2CH2CH3), via “click chemistry” under the catalysis of CuI. Those polymers were characterised using infrared spectroscopy (IR), scanning electron microscopy (SEM), and thermal gravimetric analysis (TGA). Film electrodes were assembled using a spin-coating technique by casting a mixture of the functionalised polymer, MWCNTs (multi-wall carbon nanotubes), and Nafion onto the surface of a vitreous carbon electrode. The assembled electrodes exhibited electrochemical responses and catalysis on proton reduction in a medium of acetonitrile-acetic acid with a positive shift in reduction potential by over 400 mV compared to the precursor diiron complexes (1 and 2).
CH)2(CO)6] (1) and [Fe2(μ-SnBut)(μ-SCH2CCH)(CO)6] (2, nBut = –CH2CH2CH2CH3), via “click chemistry” under the catalysis of CuI. Those polymers were characterised using infrared spectroscopy (IR), scanning electron microscopy (SEM), and thermal gravimetric analysis (TGA). Film electrodes were assembled using a spin-coating technique by casting a mixture of the functionalised polymer, MWCNTs (multi-wall carbon nanotubes), and Nafion onto the surface of a vitreous carbon electrode. The assembled electrodes exhibited electrochemical responses and catalysis on proton reduction in a medium of acetonitrile-acetic acid with a positive shift in reduction potential by over 400 mV compared to the precursor diiron complexes (1 and 2).
Introduction
It is a viewpoint widely accepted that dihydrogen may be one of the most important energy vectors in the future to replace the fossil fuels we are currently using. This is not only due to its cleanness in its combustion/oxidation from which energy is released without any by-product but water, but also its renewability and sustainability in the principle of photosynthesis. Therefore, to develop dihydrogen as our main energy source to drive our economy in the future could be one of the possible ways out of the energy crisis mankind might face sooner or later. But one of the challenges to be encountered in the course of developing the so called “hydrogen economy” is how to efficiently produce this light gas with an affordable cost. Electrolysing water to produce dihydrogen has been the major methodology in industry today in addition to reforming fossil fuels.1 The former is particularly a way to “store” the surplus electricity at an off-peak period. This would also be a solution to effectively utilise the electricity supply with the characteristic of significant fluctuation in generation, for example, the electricity generated from wind power. One of the key factors here is a catalyst. Currently, the most efficient catalysts are precious metals, platinum and palladium. There is no doubt that relying on catalysts of this kind for dihydrogen production would compromise the sustainability the dihydrogen energy vector is supposedly to have and therefore, not the solution in the long run. Thus, exploring cheaper catalysts which could be made from abundant sources has pivotal significance. In this regard, nature is apparently far smarter than our mankind.In many anaerobes, there is a family of metalloenzymes known as hydrogenase, which, to date, are classified into three types, [FeFe]–, [NiFe]–, and [Fe]–hydrogenases. Of the three enzymes, [FeFe]– and [NiFe]–hydrogenases show remarkable capabilities in both catalytic oxidation and production of dihydrogen,2 of which [FeFe]–hydrogenase has a high preference towards dihydrogen production.3,4 The relevance of dihydrogen for a future clean energy source and nature's delicate design of the system with high efficiency and rapid rate in catalysing both evolution and oxidation of dihydrogen using abundant elements in the earth's crust, have greatly inspired synthetic chemists. The revelations of the detailed structures of this enzyme over a decade ago (Fig. 1),5,6 has, indeed, prompted many chemists working on modelling the diiron sub-unit of the enzyme. Consequently, a category of classic dinuclear organometallic complexes of the core, {Fe2(μ-S)2(CO)6 − n} (n = 1 or 2 and S is not necessary an inorganic sulphide), has re-captured chemists' attention and been intensively investigated in the past decade. Recent reviews have well documented the achievements.7–9 The research has greatly advanced our understanding of the chemistry of the dinuclear complexes and hence shed light on the catalytic mechanism of the enzyme. For example, the study of a diiron pentacarbonyl complex settled finally down a controversy about the oxidation state of the distal iron of the diiron sub-unit (Fig. 1) of the enzyme at its rest state.10 Investigations into the electrochemistry of the diiron complexes finally mapped out the ECE mechanism, two-electron reduction coupled to a chemical reaction, which had not been conclusive until detailed mechanistic investigations were performed in recent years.11–14 Varying the bridging head which joins the two iron atoms together and introducing a non-CO ligand via substitution in the coordinating sphere around the dimetallic core resulted in a large number of diiron models,7–9 some of which nicely mimicked the structural features found in the enzyme.15–18
![Schematic view of the H-cluster (X = NH, CH2, or O) surrounded by protein domains in the [FeFe]–hydrogenase.](/image/article/2011/RA/c1ra00343g/c1ra00343g-f1.gif) | ||
| Fig. 1 Schematic view of the H-cluster (X = NH, CH2, or O) surrounded by protein domains in the [FeFe]–hydrogenase. | ||
On the other hand, it is known that the metal centre in the enzyme is delicately “wrapped” up by protein domains, which not only provides a “shelter” to harbour the active centre, but also a protective micro-environment to shield off exotic attacks from its surroundings and thus maintain the integrity of the active centre which is otherwise fragile. Compared to synthesising models of the diiron sub-unit alone, it is synthetically extremely challenging to assemble artificial systems of those features of the enzyme. But it is desirable to do so in the point of view of both mimicking the micro-environment composed by the protein domains and offering a “handle” in fabricating an electrode based on a synthetic model. But to date, synthetic systems of this type and reports in fabrication of catalytic electrodes using synthetic analogues have been rare. There have been two approaches of fabricating electrodes using synthetic analogues reported in the literature19–22 and models bearing carboxylic acid could be “amenable to surface immobilisation”.23 Apparently, controlling the loading of the metallic centre onto the surface of a matrix electrode has still been problematic. Recently, we reported two strategies in an attempt to tackle this issue. The two strategies involved polymerisation of the diiron model complexes armed with alkynyl group(s) and “click chemistry” between an alkynyl group and an organic azide, respectively.24–26 Although the strategies offered options to meet the challenge aforementioned, intense synthesis involved in the preparation implies that those approaches would not be cost-efficient. Therefore, in the long run, a material which is commercially available, cheap, and ready for further functionalisation is highly desirable.
PVC is such a suitable polymer to meet the criteria in this regard since it is a well known and readily available polymer with wide applications in industry.27 This material enjoys many advantages, such as high thermal and chemical stability, good film-forming properties, and decent solubility in organic solvents, for example, tetrahydrofuran (THF). More importantly, the C–Cl bonds on the backbone of PVC open a gate for further functionalisation via substitution reactions. This feature can be manipulated in the preparation of materials functionalised with diiron–carbonyl units. In fact, it is a general approach to synthesise diiron model complexes of the type, [Fe2(μ-SCH2R)2(CO)6] or [Fe2(μ-SCH2)2R′(CO)6], via reacting a diiron hexacarbonyl anion, [Fe2(μ-S−)2(CO)6], with a halide (RCH2X or R′(CH2X)2, both R and R′ are organic moieties).28 This well established approach and the aforementioned advantages possessed by PVC, as well as our recent success in assembling an electrode using “click chemistry”,24–26 prompted us to explore the chloro-functionality of this polymer in our continual efforts to explore artificial systems suitable for assembling film electrodes.
Herein, we report the preparations of three PVC-based polymeric materials functionalised with the diiron units, {Fe2(CO)6 or 5}, PVC–Fe-A, -B, and -C, their assembly of film electrodes, and electrochemical investigations. The polymer PVC–Fe-A was prepared through direct reaction between PVC and the reduced form of the complex, [Fe2(μ-S)2(CO)6], while the other two were prepared using “click chemistry” after PVC had been functionalised with azide groups. These functionalised polymers were characterised using IR, TGA, and SEM techniques. Into those polymers were blended with MWCNTs and Nafion to improve the conductivity and film-forming property of the materials. The film electrodes were assembled via spin-coating the blend onto the surface of a vitreous carbon electrode. Electrochemical behaviours of the film electrodes were also investigated using cyclic voltammetry.
Experimental
Materials, instrumentation, and general procedures
Unless otherwise indicated, most of the reactions were conducted using standard Schlenk techniques under an Ar atmosphere. Solvents were dried and distilled appropriately prior to use. Infrared spectral data were collected on a Scimitar 2000 (Varian). NMR spectra were recorded on either an Avance DRX 400 or Bruker Advance 600 (Bruker). Thermal gravimetric analysis measurements were evaluated with a TA SDT Q600 at a heating rate of 10 °C min−1 under nitrogen flow. PVC (MW = 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma-Aldrich. Complexes, [Fe2(μ-S)2(CO)6], [Fe2(μ-SCH2CCH)2(CO)6] (1) and [Fe2(μ-SnBu)(μ-SCH2CCH)(CO)6] (2) were synthesised following literature procedures and synthetic details were described in our recent publications.23,25
000) was purchased from Sigma-Aldrich. Complexes, [Fe2(μ-S)2(CO)6], [Fe2(μ-SCH2CCH)2(CO)6] (1) and [Fe2(μ-SnBu)(μ-SCH2CCH)(CO)6] (2) were synthesised following literature procedures and synthetic details were described in our recent publications.23,25
Synthesis
CH in toluene (0.7 mL, 6.3 mmol) was added dropwise. The reaction was then allowed to slowly warm to room temperature to produce a red solution before further stirring for 2 h. Removal of the solvents from the solution gave a dark-red residue. Purification of the residue using flash chromatography (eluent: petroleum ether) produced a red liquid. Recrystallisation of this liquid in hexanes gave a red solid (0.398 g, 30%). IR (hexane, cm−1): υCO = 2078 (m), 2044 (s), 2009 (m), 1999 (m); υCCH = 3316 (w).
Complex 2: to a pre-cooled solution of [Fe2(μ-S)2(CO)6] (1.72 g, 5 mmol) in THF (60 mL, −78 °C in a dry-ice–acetone bath) nBuLi (2.3 mL, 2.2 mol L−1) was slowly added over 15 min. The reaction mixture turned from red to dark green upon the addition. After being stirred for 20 min, a solution of BrCH2CCH in toluene (0.6 mL, 5.4 mmol) was added dropwise to the reaction, before the reaction mixture was allowed to slowly warm to room temperature which gave a red solution. The reaction was further stirred for 1 h before removal of the solvent from the red solution which produced a dark liquid. Purification of the crude product with flash chromatography (eluent: petroleum ether) gave a red liquid (0.792 g, 36%). IR (hexane, cm−1): υCO = 2075 (m), 2040 (s), 2005 (m) and 1995 (m); υCCH = 3316 (w).
PVC–N3. PVC (1.25 g, 20 mmol) and NaN3 (0.39 g, 6 mmol) was added into DMF (20 mL). The mixture was stirred at 30 °C for 2 days and then poured into water (400 mL) at room temperature to precipitate the product which was collected via filtration. The collected solid was redissolved in THF (10 mL) and the solution was added dropwise to methanol (500 mL) under stirring to precipitate the functionalised polymer. The mixture was left to stand for 30 min to allow the polymer to settle down before it was collected using filtration. The collected polymer was washed with methanol (3 × 20 mL) and dried to a constant weight (light yellow solid, 1.09 g). IR (KBr pellets, cm−1): 2113 (azide).
PVC–Fe-A. A solution of [Fe2(μ-S)2(CO)6] (516 mg, 1.5 mmol) in THF (40 mL) was cooled to −78 °C using a dry-ice–acetone bath. To the solution LiHBEt3 (3.0 mL, 1.0 mol L−1) was added with stirring and under an Ar atmosphere. The reaction mixture turned from red to dark green upon the addition. After being stirred for 15 min, the green solution was added to a solution of PVC (375 mg, 6 mmol) in THF (20 mL) through a cannula. The reaction mixture was allowed to slowly warm to room temperature and then heated at 40 °C for 16 h before CH3I (0.3 mL) was added. Upon the addition, the reaction changed shortly to red. The mixture was maintained at the same temperature with stirring for a further 6 h. After removal of any insoluble material, the filtrate was concentrated to about 8 mL which was then added dropwise to methanol (500 mL) under stirring. A precipitate formed in the mixture upon the addition. After leaving to stand for 2 h, the precipitate was filtered, washed with methanol (3 × 10 mL), and dried in vacuum to a constant weight (a dull-yellow solid, 386 mg). IR (KBr pellets, υCO, cm−1): 2072 (m), 2034 (s), 1991 (m).
PVC–Fe-B. PVC–N3 (260 mg) and complex 1 (334 mg, 0.76 mmol) were dissolved in dry THF (40 mL) in a Schlenk flask (100 mL) under an Ar atmosphere. To the solution a suspension of CuI (18 mg) in THF (10 mL) was added through a cannula, which was followed by the addition of Et3N (1.0 mL). The reaction mixture was stirred at 40 °C overnight to give a dark-red solution. After removal of insoluble solids by filtration, the filtrate was concentrated to about 8 mL under a reduced pressure and then added dropwise to methanol (300 mL) under stirring to precipitate the product. After being left to stand for 30 min, the precipitate was collected via filtration, washed with methanol (3 × 10 mL), and finally dried in vacuum to a constant weight (deep brown solid, 310 mg). IR (KBr pellets, υCO, cm−1): 2070 (m), 2033 (s), 1988 (m), 1951(m); 2115 (s, azide).
PVC–Fe-C. The polymer was prepared as a red solid by reacting PVC–N3 with complex 2 in a manner analogous to the preparation of PVC–Fe-B. IR (KBr pellets, υCO, cm−1), 2074 (m), 2038 (s), 1992 (m), 1963 (m); 2113 (s, azide).
Fabrication of film electrodes and electrochemical investigation
A film electrode was achieved by spin-coating a mixture containing the prepared functionalised polymer suspended in Nafion solution (10 mg mL−1) and an appropriate amount of MWCNTs (please refer to the ESI for further details†) onto the surface of a vitreous electrode. The coated film electrode was left to dry in an Ar flow. Electrochemistry was performed in a self-designed gas-tight cell equipped with three-electrodes under Ar and at 298 K on an Autolab PGSTAT 30. All potentials were quoted against the Fc+/Fc couple. Vitreous carbon disk (φ = 5 mm) was used as the working electrode when the film electrode was fabricated, but an electrode of 1 mm in diameter was used for the electrochemistry of complexes 1 and 2. The electrochemical cell set-up for electrochemistry is generally the same as those described elsewhere in our recent work.13,29,30Results and discussion
Preparation of the functionalised polymers
In this work, the reaction between the reduced form of a classic diiron hexacarbonyl complex, [Fe2(μ-S)2(CO)6], and an organic chloride was exploited to prepare our first PVC-based functionalised polymer, Scheme 1. As shown in Scheme 1, it is feasible to think of two manners for the functionalisation to proceed. In one way, every two adjacent reaction sites (C–Cl) along the polymer backbone react with one diiron complex to give form A, and in the other way, cross-linking between the polymer chains may occur to produce polymer form B. In either case, the reaction should show the characteristic dark-red colour possessed by the diiron hexacarbonyl complexes. In fact, the reaction solution remained an intense green, the colour largely from the reduced diiron precursor. But addition of PVC into the reaction did shift the spectral profile towards higher frequencies, which strongly suggested that reaction occurred but full substitution of the reduced diiron hexacarbonyl units with PVC was not yet achieved. Further addition of CH3I into the reaction brought the spectral absorption bands to further higher frequencies comparable to those for the diiron hexacarbonyl complexes. The stepwise spectral variation is shown in Fig. 2. Addition of CH3I into the solution led to the isolation of [Fe2(μ-SCH3)2(CO)6]33 and functionalised polymer (PVC–Fe-A) soluble in THF. Fig. 3 shows the infrared spectra of the polymer, the precursor, and diiron by-product. Thus, a full scenario of the substitution reaction can essentially be established based on the experimental observations and above arguments.
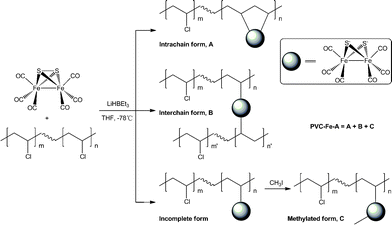 | ||
| Scheme 1 Synthetic route for the preparation of PVC–Fe-A and its possible forms. | ||
![Spectral variation during the reaction course (reaction solvent: THF). Please note that except for the precursor, [Fe2(μ-SCH3)2(CO)6], unknown species existed in the reaction solutions in addition to the principle component as discussed in the text.](/image/article/2011/RA/c1ra00343g/c1ra00343g-f2.gif) | ||
| Fig. 2 Spectral variation during the reaction course (reaction solvent: THF). Please note that except for the precursor, [Fe2(μ-SCH3)2(CO)6], unknown species existed in the reaction solutions in addition to the principle component as discussed in the text. | ||
![Infrared spectra of the precursor (dashed line, in THF), [Fe2(μ-S)2(CO)6], the by-product (dotted line, in CCl4), [Fe2(μ-SCH3)2(CO)6], and PVC–Fe-A (solid line, in THF).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f3.gif) | ||
| Fig. 3 Infrared spectra of the precursor (dashed line, in THF), [Fe2(μ-S)2(CO)6], the by-product (dotted line, in CCl4), [Fe2(μ-SCH3)2(CO)6], and PVC–Fe-A (solid line, in THF). | ||
Upon the addition of PVC into the solution of the reduced precursor, the intrachain and interchain cross-linking reactions may occur to give the products, forms A and B, respectively. But experimental observations suggested that such reactions are beyond infrared spectral detection since the addition of PVC exhibited no noticeable spectral characteristic for any diiron hexacarbonyl unit which both forms A and B should possess. This incomplete substitution reaction could be solely attributed to the steric effect. The real situation could be as follows. After the addition of PVC into the solution of the reduced diiron precursor, some of it was “half-reacted”, that is, only one of the two reduced bound thiolates of each diiron unit reacted with PVC and left the other one intact, and the rest were unreacted. Addition of CH3I quenched the reaction to complete the alkylation of the reduced diiron unit to give both [Fe2(μ-SCH3)2(CO)6] (not shown) and product form C, Scheme 1. The scenario described above was further confirmed by employing 3-chloropropanoyl chloride (ClCH2CH2COCl) in quenching the reaction as the resultant analogous polymer is more informative due to the infrared spectroscopic “finger-print” of the functional group (thioester) in the functionalised polymer (Fig. S1†). The analogous polymer shows clearly an absorption band at 1779 cm−1 which is attributed to the thioester carbonyl (–SCOCH2CH2Cl) bound to the diiron units.
 | ||
| Scheme 2 Preparation and schematic views of polymers PVC–N3 and PVC–Fe-B, and possible conformations of PVC–Fe-B. | ||
When complex 2 which possesses only one alkynyl group was employed, the cross-linking reaction which occurred in the preparation of polymer PVC–Fe-B was completely eliminated. Thus the resultant functionalised polymer, PVC–Fe-C, has the sole form as depicted in Scheme 3. Elimination of the interchain cross-linking renders the polymer good solubility in most common solvents such as THF and thus the best film-forming property among the three polymers. It has to be noted that both PVC–Fe-B and -C possess a significant amount of unreacted azides as indicated by their infrared spectra, Fig. 4. The minor absorption band right after the azide band is attributed to the terminal hexacarbonyl diiron unit (vide infra).
 | ||
| Fig. 4 Infrared spectra of the polymers (KBr pellets). | ||
 | ||
| Scheme 3 Preparation and schematic view of polymer PVC–Fe-C. | ||
The coordination chemistry of the diiron units in the polymers, their thermal stability, and morphology of the polymers
It is well known that complexes of the diiron units, {Fe2(CO)n} (n = 4–6) show a characteristic spectral pattern. Thus, examining their infrared spectral profiles surrenders the most useful structural information of the diiron units in the polymers. The infrared spectra related to the bound CO are shown in Fig. 4. It is noteworthy that in the spectra of PVC–N3, PVC–Fe-B, and PVC–Fe-C, the prominent band at just above 2100 cm−1 is attributed to the azide group as mentioned above. As indicated by the spectral profiles shown in Fig. 4, PVC–Fe-A and the other two polymers (PVC–Fe-B and PVC–Fe-C) fall apparently into two categories, one of which possesses the {Fe2(CO)6} unit, and the other category consists of PVC–Fe-B and PVC–Fe-C in which the diiron unit is of the type, {Fe2(CO)5. The latter coordination chemistry is consistent with our results reported recently.25 In the two polymers, one of the three N atoms of the triazol ring formed via “click chemistry” coordinates to the diiron unit (Scheme 2 and 3). As mentioned above, the latter two polymers have a minor absorption band at roughly the same position as that of the highest absorption band of PVC–Fe-A in addition to the major bands. This is due to the terminal diiron unit which does not have a triazol ring to bind and therefore, is a diiron hexacarbonyl unit.The thermal stability of the polymers was evaluated using thermal gravimetric analysis (TGA) between 25 °C and 750 °C under a steady stream of dry nitrogen. All the TGA diagrams of the polymers are shown in Fig. 5. PVC exhibits two distinct stages of thermal decompositions, that is, evolution of HCl first and then the destruction of its carbon backbone.34 The observed weight-loss of about 60% as shown in Fig. 5 is rather close to the calculated 55%. Functionalisation of PVC generally decreases the thermal stability. For PVC–N3, except for the shift of the TGA trace of the first stage due to the decrease in decomposition temperature by approximately 80 °C, the entire profile is largely the same as that of PVC. This may suggest that the substitution of chloride by azide is quite complete.
 | ||
| Fig. 5 TGA traces of the polymers (inset: the zoomed-in view of the initial stages of the thermal decomposition of the polymers). | ||
Immobilisation of diiron units onto the PVC chain further weakens the thermal stability of the polymers. Compared to the commencing temperature of decomposition of PVC, the temperature lowered by ca. 150 °C. On the other hand, all the functionalised polymers showed a better thermal stability by about 100 °C compared to the analogous polymers we reported recently.24,25 This improvement is certainly associated with the 3-D structures of the polymers which could detain the thermal destruction of the iron carbonyl units in the polymers.
The organic polymers (PVC and PVC–N3) give very neat two-stage decompositions and leave ca. 10% residue after thermal decomposition (Fig. 5). For the iron-containing polymers, this residue increases by about 25%. By assuming that the residue is consisted of Fe–S compounds, the CO-loss in total accounts for approximately 20%, which is about two-folds the loss at the first stage (<ca. 450 K). This suggests that the CO-loss due to thermal decomposition is not completed at the first stage.
Close examination of the TGA traces of the three polymers (Fig. 5, inset) reveals that PVC–Fe-A is different from the other two polymers. For PVC–Fe-A, its thermal stability is poorer. Its thermal decomposition commences almost no sooner than the heating starts. This may be due to the greater steric tension compared to the other polymers. This tension originates from the attaching manner with which the diiron units are directly linked to the polymer backbone via C–S bonds, Scheme 1. When the temperature reaches just about 500 K, its weight-loss is over 30%, of which about 10% is attributed to CO-loss. The rest ought to account for Cl-related weight-loss since there is certainly some chloro-backbone intact in this polymer but interestingly, the thermal decomposition temperature decreases by about 50 °C. For polymers PVC–Fe-B and PVC–Fe-C, there is an extra transient stage between 550 K and 650 K in addition to those observed for PVC–Fe-A. This is probably from the triazol-related thermal decomposition.
As indicated by the SEM images of the polymers (Fig. 6, A–E), from PVC to the functionalised polymers, there are significant changes in morphology. In general, the good film-forming property of PVC deteriorates with attachment of extra groups onto the polymer backbone by substitution of the chloride. When the group (azide) is not significantly different from the chloride, the polymer (PVC–N3) adopts beautiful spherical shapes at the scales of hundred nanometres. This spherical shape was replaced with irregular congregates when the diiron units were chemically grafted onto the backbone.
 | ||
| Fig. 6 SEM images of the functionalised polymers (the scale bar in each image indicating 500 nm) and A: PVC, B: PVC–N3, C–E: PVC–Fe-A, -B and -C, respectively, F–H: composites of PVC–Fe-A, -B and -C, respectively, with MWCNTs and Nafion. | ||
3.3 Assembling film electrodes with the functionalised polymers and their electrochemical investigations
As discussed above, by exploiting the chloride functionality of PVC, diiron carbonyl units can be attached to the backbone of the polymer to generate the three polymers, PVC–Fe-A, -B, and -C. It is our long term goal to look for an appropriate polymer as a matrix to hold the bio-inspired diiron models, assemble artificial systems into film electrodes. To examine these polymers in this regard, there are two obstacles to overcome, (i) these polymers are not conductive, and (ii) the film-forming property of these polymers altered due to incorporation of the diiron models onto the polymer backbone. Particularly, polymer PVC–Fe-B is poorly soluble in common solvents due to the severe cross-linking reaction which occurred in the functionalisation. Even for the other two polymers which are soluble in THF, relying entirely on doping MWCNTs in their THF solutions generated no satisfactory results (data not shown). Thus, a strategy of combining both Nafion and MWCNTs was employed in assembling film electrodes with the three polymers. Nafion not only enhances the film-forming property of these polymers, but also offer an improvement in conductivity. The addition of MWCNTs further improves the conductivity of the film as suggested by our data shown below. Fig. 5 (F–H) shows the SEM images of the films formed with the three polymers mixed with an appropriate amount of Nafion and MWCNTs. The images suggest that the components are well dispersed in the composites under supersonification. With the composites, film electrodes were prepared via spin-coating onto the surface of a vitreous carbon electrode (typically φ = 5 mm). The thickness of the film is controlled at the micrometre scale which was approximately between 10–30 μm as estimated using SEM.Electrochemical behaviours of the film electrodes assembled using the three functionalised polymers are shown in Fig. 7, 8, and 9, respectively. Compared to the monomer diiron models (1 and 2), the film electrodes in acetonitrile solution show reduction process more positively by over 100 mV, Fig. 8 and 9. By using Nafion and PVC–N3–Nafion, respectively, as a control, it is clear that the observed reduction is {FeFe}-based. The results also reveal the enhancement of MWCNTs towards the electrochemical responses. The improvement in reduction current is due to the increase in conductivity by the added MWCNTs. The positive shift in reduction potential is attributed to the polymeric nature of the systems.24,25
![Cyclic voltammograms of monomer 1 (C = 2.9 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-A–Nafion, PVC–Fe-A–MWCNTs–Nafion on the vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f7.gif) | ||
| Fig. 7 Cyclic voltammograms of monomer 1 (C = 2.9 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-A–Nafion, PVC–Fe-A–MWCNTs–Nafion on the vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K). | ||
![Cyclic voltammograms of monomer 1 (C = 2.9 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-B–Nafion, PVC–Fe-B–MWCNTs–Nafion on the vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1(298 K).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f8.gif) | ||
| Fig. 8 Cyclic voltammograms of monomer 1 (C = 2.9 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-B–Nafion, PVC–Fe-B–MWCNTs–Nafion on the vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1(298 K). | ||
![Cyclic voltammograms of monomer 2 (C = 6.6 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-C–Nafion, PVC–Fe-C–MWCNTs–Nafion on the surface of a vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f9.gif) | ||
| Fig. 9 Cyclic voltammograms of monomer 2 (C = 6.6 mmol L−1), Nafion, PVC–N3–Nafion, PVC–Fe-C–Nafion, PVC–Fe-C–MWCNTs–Nafion on the surface of a vitreous carbon electrode in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K). | ||
It is well established that diiron complexes of the core, {Fe2(CO)4–6}, in which the two iron atoms are usually bridged by two thiolates, undergo a two-electron process via an ECE mechanism.12–14,35,36 The feature that the second electron transfer after the coupled chemical reaction occurs often at a potential more positive than the first one renders the reduction process appearing as a single-wave. In the coupled chemical reaction, the diiron core and its surrounding ligands undergo reorganisation or isomerisation to generate a species which, compared to the potential for the first one-electron reduction, accepts the second electron at a more positive or slightly more negative potential due to its thermal instability. But in the film, the coupled chemical reaction should not be the same as that in solution chemistry due to the confinement of the diiron units imposed by the 3-D conformation of the polymers. It is noticeable that there is a minor reduction at ca. −1.8 V in both cases as shown in Fig. 8 and 9. Close examination of this process suggests that the weak process may be assigned to the proton reduction associated with the Nafion in the film. For both monomers 1 and 2, they show essentially a single-wave reduction at 1.47 V and 1.55 V, respectively.
Our results indicate that the commercially available material PVC can be used as a matrix to incorporate the diiron units. The applications of Nafion and MWCNTs ensure the functionalised polymers electrochemical activity and feasibility of assembling into a film electrode via spin-coating. But further concerns are how durable the film electrodes are, and whether the electrodes respond to the addition of a weak acid, for example, acetic acid. To address the first concern, repetitive scanning of the film electrode was performed through an appropriate potential range between which the reductions occur. Fig. 10 shows the results of repetitive scanning of the film electrode made from polymer PVC–Fe-C. After the first scan, the current drops by ca. 80%, which indicates that reductive bleaching-off of the diiron units in the film exists on one hand. On the other hand, the loss of the reduction current can not be entirely attributed to the loss of electrochemically active components in the film since infrared spectroscopic examination after 30 repetitive scans indicated only about 60% loss of the carbonyl-containing components (Fig. S2 and S3†). Of the two experimental observations, the infrared spectroscopic one may be more informative than the electrochemical one as the internal structure and conductivity may be altered quite significantly after the first scan. The alteration would have a direct influence on the electrochemical response of the film electrode, and have less of an influence on the infrared spectroscopic measurement. These results suggest that to improve the durability of the film electrodes, employing diiron units possessing resistance against reductive destruction is necessary, and on the other hand, incorporating components positively charged into the film may be helpful in stabilising the reduced diiron units.
![Cyclic voltammograms of the film electrode prepared from PVC–Fe-C–MWCNTs–Nafion after repetitive scanning (1st, 2nd, 3rd, 6th, and 9th from the bottom to the top) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scanning rate of 0.1 V s−1 (298 K).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f10.gif) | ||
| Fig. 10 Cyclic voltammograms of the film electrode prepared from PVC–Fe-C–MWCNTs–Nafion after repetitive scanning (1st, 2nd, 3rd, 6th, and 9th from the bottom to the top) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scanning rate of 0.1 V s−1 (298 K). | ||
Since the electrochemical components in the film electrode are bleached off with scanning, a number of film electrodes assembled under the same conditions were used to assess how the film electrodes respond to the addition of acetic acid. For the film electrodes prepared using the three functionalised polymers, their responses upon acid addition are shown in Fig. 11, 12, and 13, respectively.
![Electrochemical responses of the film electrodes (PVC–Fe-A–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 at 298 K.](/image/article/2011/RA/c1ra00343g/c1ra00343g-f11.gif) | ||
| Fig. 11 Electrochemical responses of the film electrodes (PVC–Fe-A–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 at 298 K. | ||
![Electrochemical responses of the film electrodes (PVC–Fe-B–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K).](/image/article/2011/RA/c1ra00343g/c1ra00343g-f12.gif) | ||
| Fig. 12 Electrochemical responses of the film electrodes (PVC–Fe-B–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 (298 K). | ||
![Electrochemical responses of the film electrodes (PVC–Fe-C–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 at 298 K.](/image/article/2011/RA/c1ra00343g/c1ra00343g-f13.gif) | ||
| Fig. 13 Electrochemical responses of the film electrodes (PVC–Fe-C–MWCNTs–Nafion) at various concentrations of acetic acid (HOAc) in 0.1 mol L−1 [NBu4]BF4–CH3CN solution at a scan rate of 0.1 V s−1 at 298 K. | ||
There has been the view that for complexes of diiron carbonyl in solution electrochemistry, the species from the one-electron reduction is not responsible for catalytic reduction of a proton and instead species from further reduction of the monoanion after protonation performs the catalysis.12,13,36–38 This protonated species is supposedly at first a terminal hydride since it is known that a bridging hydride does not show catalytic reduction towards proton reduction under similar conditions even when it is further reduced.39–41 But the terminal hydride is unstable and transforms into its bridging form.12,40,41 Probably, the catalytic efficiency is associated with the transforming rate. In the film electrode, the confinement of the catalytic species imposed by the polymer is certainly beneficial in this regard. But this confinement may also increase resistance for proton transportation in the catalysis and thus affect the catalytic efficiency. As in their electrochemical responses without the presence of the acid, after the major catalytic peak, there is a minor reduction process as well. But no catalysis was observed for this minor reduction process, which further strengthens our assignment made earlier that this process is likely to have originated from the proton reduction related to the Nafion.
Conclusions
Exploitation of the C–Cl functionality of the widely available commercial plastic, PVC, led to three functional polymers, PVC–Fe-A, -B, and -C. The direct reaction of the reduced diiron precursor, [Fe2(μ-S−)2(CO)6], with the plastic produced polymer PVC–Fe-A while two other functionalised polymers (PVC–Fe-B and -C) were derived via “click chemistry” between the alkynyl group of the diiron models and the azide group grafted chemically on the PVC backbone. Blending the functionalised polymers with MWCNTs and Nafion enables the assembly of film electrodes which show electrochemical responses originating from the incorporated diiron units and catalysis on proton reduction in the medium of acetonitrile-acetic acid. Due to the polymeric effect,24,25 the potential for the catalysis of proton reduction shifted positively over 400 mV compared to that of monomer diiron complexes.Our results show that the commercially available PVC polymer can be a matrix to hold the diiron model units. However, achieving a system for assembling robust film electrodes poses still a great challenge. The points that need to be further addressed are the resistance of the incorporated diiron units against reductive destruction, the 3-D structure of the functionalised polymers and the properties of the assembled films. In the end, the improvements in these aspects are desired to ensure better stability of the reduced diiron units and such a film that the metallic centres are stabilised upon reduction, electron/proton transfer are enhanced, and hydrophilicity is exhibited for operation in aqueous solution.
Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 20871064), the Ministry of Science and Technology (China) (973 programs, Grant No. 2009CB220009). The provincial government of Zhejiang is also acknowledged for the Qianjiang Professorship (XL) at Jiaxing University.References
- U.S. Department of Energy Hydrogen Program, http://www.hydrogen.energy.gov/pdfs/doe_h2_production.pdf .
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS.
- Y. Nicolet, B. J. Lemon, J. C. Fontecilla-Camps and J. W. Peters, Trends Biochem. Sci., 2000, 25, 138–143 CrossRef CAS.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- X. M. Liu, S. K. Ibrahim, C. Tard and C. J. Pickett, Coord. Chem. Rev., 2005, 249, 1641–1652 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, Curr. Opin. Biotechnol., 2010, 21, 292–297 CrossRef CAS.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–275 RSC.
- G. A. N. Felton, B. J. Petro, R. S. Glass, D. L. Lichtenberger and D. H. Evans, J. Am. Chem. Soc., 2009, 131, 11290–11291 CrossRef CAS.
- J. F. Capon, F. Gloaguen, F. Y. Petillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2009, 253, 1476–1494 CrossRef CAS.
- Y. W. Wang, Z. M. Li, X. H. Zeng, X. F. Wang, C. X. Zhan, Y. Q. Liu, X. R. Zeng, Q. Y. Luo and X. M. Liu, New J. Chem., 2009, 33, 1780–1789 RSC.
- X. H. Zeng, Z. M. Li, Z. Y. Xiao, Y. W. Wang and X. M. Liu, Electrochem. Commun., 2010, 12, 342–345 CrossRef CAS.
- C. Tard, X. M. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L. S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS.
- J. I. van der Vlugt, T. B. Rauchfuss and S. R. Wilson, Chem.–Eur. J., 2006, 12, 90–98 CrossRef.
- T. Liu and M. Y. Darensbourg, J. Am. Chem. Soc., 2007, 129, 7008–7009 CrossRef CAS.
- C. M. Thomas, T. Liu, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2008, 47, 7009–7024 CrossRef CAS.
- V. Vijaikanth, J. F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Electrochem. Commun., 2005, 7, 427–430 CrossRef CAS.
- S. K. Ibrahim, X. M. Liu, C. Tard and C. J. Pickett, Chem. Commun., 2007, 1535–1537 RSC.
- S. Ibrahim, P. M. Woi, Y. Alias and C. J. Pickett, Chem. Commun., 2010, 46, 8189–8191 RSC.
- A. Le Goff, V. Artero, R. Metaye, F. Moggia, B. Jousselme, M. Razavet, P. D. Tran, S. Palacin and M. Fontecave, Int. J. Hydrogen Energy, 2010, 35, 10790–10796 CrossRef CAS.
- C. M. Thomas, O. Rüdiger, T. Liu, C. E. Carson, M. B. Hall and M. Y. Darensbourg, Organometallics, 2007, 26, 3976–3984 CrossRef CAS.
- X. Ru, X. H. Zeng, Z. M. Li, D. J. Evans, C. X. Zhan, Y. Tang, L. J. Wang and X. M. Liu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2410–2417 CrossRef CAS.
- C. X. Zhan, X. F. Wang, Z. H. Wei, D. J. Evans, X. A. Ru, X. H. Zeng and X. M. Liu, Dalton Trans., 2010, 39, 11255–11262 RSC.
- X. M. Liu, X. Ru, Y. Li, K. K. Zhang and D. Y. Chen, Int. J. Hydrogen Energy, 2011, 36, 9612–9619 CrossRef CAS.
- R. H. Burgess, Manufacture and processing of PVC, Applied Science, London, 1982 Search PubMed.
- J. D. Lawrence, H. X. Li and T. B. Rauchfuss, Chem. Commun., 2001, 1482–1483 RSC.
- Z. M. Li, X. H. Zeng, Z. G. Niu and X. M. Liu, Electrochim. Acta, 2009, 54, 3638–3644 CrossRef CAS.
- X. H. Zeng, Z. M. Li and X. M. Liu, Electrochim. Acta, 2010, 55, 2179–2185 CrossRef CAS.
- T. Kameda, Y. Fukuda, G. Grause and T. Yoshioka, J. Appl. Polym. Sci., 2010, 116, 36–44 CrossRef CAS.
- M. Y. Abdelaal and T. R. Sobahi, J. Appl. Polym. Sci., 2007, 104, 2304–2309 CrossRef CAS.
- D. Seyferth, R. S. Henderson and L. C. Song, Organometallics, 1982, 1, 125–133 CrossRef CAS.
- R. Font, A. Galvez, J. Molto, A. Fullana and I. Aracil, Chemosphere, 2010, 78, 152–159 CrossRef CAS.
- Z. Y. Xiao, Z. H. Wei, L. Long, Y. L. Wang, D. J. Evans and X. M. Liu, Dalton Trans., 2011, 40, 4291–4299 RSC.
- Y. Tang, Z. H. Wei, W. Zhong and X. M. Liu, Eur. J. Inorg. Chem., 2011, 1112–1120 CrossRef CAS.
- S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. M. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988–16999 CrossRef CAS.
- S. J. Borg, S. K. Ibrahim, C. J. Pickett and S. P. Best, C. R. Chim., 2008, 11, 852–860 CrossRef CAS.
- D. Morvan, J. F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Eur. J. Inorg. Chem., 2007, 5062–5068 CrossRef CAS.
- B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2008, 47, 2261–2263 CrossRef CAS.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 16834–16835 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00343g |
| This journal is © The Royal Society of Chemistry 2011 |