Sequential crystallization of sea urchin-like bimetallic (Ni, Co) carbonate hydroxide and its morphology conserved conversion to porous NiCo2O4 spinel for pseudocapacitors†
Junwu
Xiao
and
Shihe
Yang
*
Department of Chemistry, William Mong Institute of Nano Science and Technology, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong (China). E-mail: chsyang@ust.hk
First published on 10th August 2011
Abstract
We report kinetic control over and mechanistic studies on the formation of sea urchin-like, bimetallic (Ni, Co) carbonate hydroxidevia a sequential crystallization process, which was facilely converted to porous NiCo2O4 spinel with a conserved morphology, an excellent candidate material for pseudocapacitors. The formation of bimetallic carbonate hydroxide was found to start with the nucleation of monometallic nickel carbonate hydroxide evolving into flower-like microspheres. This was followed by the nucleation and growth of the bimetallic carbonate hydroxide nanorods from and on the nanoplates in the flower-like microspheres by localized dissolution-recrystallization, leading finally to the sea urchin structure. After calcination, a morphology conserved NiCo2O4 spinel nanostructure was formed, which uniquely comprises hierarchical, interconnected pores with high specific surface areas suitable for fast electron and electrolyte transport. This, in tandem with the rich redox reactions of nickel cobaltite spinel and their at least two orders of magnitude higher electric conductivity than those of nickel oxides and cobalt oxides alone, renders the novel nanostructures ideal candidates for pseudocapacitors. Indeed, the porous NiCo2O4 nanostructure with a specific surface area of up to 198.9 m2 g−1 has exhibited higher specific capacitances (658 F g−1 at 1 A g−1) than the monometallic cobalt oxides (60 F/g at 1 A g−1) and nickel oxides (194 F g−1 at 1 A g−) with similar porous nanostructures. Significantly, even at a high current density of 10 A g−1, the pseudocapacitor made of NiCo2O4 porous materials retained high specific capacitances of 530 F g−1 with excellent cycling stability. In all, the simple, scalable syntheses and the excellent supercapacitor performance reported here portend large scale applications of these novel materials in energy storage.
Introduction
Supercapacitors can provide transient but extremely high power density, which are probably one of the most important next generation energy storage devices.1 They have important applications in hybrid electrical vehicles to provide peak power during acceleration in combination with batteries,2 and are key to the future development of mobile technology and micro-electromechanical systems. They can be divided into electrical double layer capacitors and pseudocapacitors depending on the energy storage mechanisms. Electrical double layer capacitor materials are usually carbon materials with high surface area such as carbon nanotubes, porous carbon, and graphene, since the specific capacitance relies on the electrical charge stored at the interface between and the electrode and electrolyte. However, a low specific capacitance of only around 100∼200 F g−1 is their main drawback holding back their large-scale application.Pseudocapacitors, which rely on faradic reactions, can achieve much high specific capacitance than electrical double layer capacitors. Pseudocapacitor materials possess multiple oxidation states/structures capable of rich redox reactions, which include transition metal oxides, nitrides and sulfides and conducting polymers. However, during the fast and reversible faradaic processes of pseudocapacitors, the poor electric conductivity of transition metal oxides and the mechanical degradation of conducting polymers hinder their applications as electrode materials. The most notable among them are RuO2, which can achieve a specific capacitance as high as 1340 F g−1.3 However, the sheer high cost and rareness of Ru prevent it from industrial applications. Other metal oxides, such as nickel oxide, cobalt oxide and manganese oxide, have been explored for supercapacitors to replace RuO2. In comparison to RuO2, these metal oxides, i.e., Co3O4,4,5NiO,6,7 and MnO2,8 have exhibited lower specific capacitance, primarily because they are typically too insulating to support fast electron transport required by high rate supercapacitors between electrolyte and electroactive species.
High specific capacitance of a pseudocapacitor requires numerous electroactive sites in the electrodes and high transport rates of electrolyte ions and electrons simultaneously taking part in the faradaic reactions. The former involves large specific surface areas of electroactive materials, while the later entails fast diffusion of the electrolyte ions and fast conduction of electrons to the rich electroactive sites. This can be achieved by concocting hierarchical (meso- and macro-) porosity of electroactive materials with high specific surface areas, high electrical conductivity and fast ion transport. By common knowledge, nickel cobaltite spinel possess a higher electronic conductivity, by at least two orders of magnitude, and a higher electrochemical activity, due to their richer redox reactions with contributions from both nickel and cobalt ions, than those of the monometallic nickel oxides and cobalt oxides.9 Thus, it appears that nickel cobaltite spinel, when fashioned into a hierarchical porous ilk, could be a material of choice for pseudocapacitors.
Commonly, porous structures can be obtained via sacrificial template, sol-gel and thermal decomposition methods. Using the sacrificial template method, templates can be divided into soft templates (surfactants and copolymers)10 and hard templates (porous silica and carbon, polystyrene and poly(methyl methacrylate) spheres).11,12 However, to fully remove the template is a tough process, since it needs to operate at relatively high temperatures, e.g., above 500 °C, and uses dangerous and toxic chemical agents, e.g., HF and sodium hydroxide, to remove the templates while maintaining the porous structure without collapsing. Obvious disadvantages include poor crystallinity for the soft template method and complicated synthesis for the hard templates approach.13 Next, an epoxide-driven sol-gel process, followed by drying in supercritical carbon dioxide, was used to synthesize a mixed aerogel of NiCo2O4 and Ni(OH)2.14 Although the specific surface area of the as-prepared aerogel was ∼120 m2 g−1, it decreased precipitously to 43 m2 g−1, concurrent with a dramatic decrease of the specific capacitance, after calcination at 300 °C, a process needed to stabilize the materials framework.
As to the thermal decomposition process, one first co-precipitate metal cations with carbonate and/or hydroxide anions to form metal carbonate and/or hydroxide precursors, followed by thermal decomposition to form porous metal oxides at relatively low temperatures accompanied by the release of CO2 and water vapor. In comparison with the sacrificial template method, the thermal decomposition method is much simpler and particularly apposite for preparing porous transition metal oxide nanostructures because of the availability of many different appropriate precursors. For example, porous nickel cobaltite spinel has been obtained by thermally decomposing carbonate and/or hydroxide precursors; but the specific surface area was only 90 m2 g−1 and there were no obvious nanostructures due to the uncontrollable co-precipitation method used.15 Herein, we report our successful synthesis of sea urchin-like porous nickel cobaltite spinel nanostructures by thermal decomposition of appropriate carbonate hydroxide precursors but without the calcination-induced framework instability problem mentioned above. The key to forming the nanostructures is to control the precipitation of the corresponding carbonate hydroxide precursors through a slow supply of the carbonate and hydroxide anions from hydrolysis of urea. We found that the sea urchin-like NiCo2O4 spinel porous materials, with specific surface areas of 198.9 m2 g−1, have demonstrated the high specific capacitances of 658 F g−1, respectively, at a current density of 1 A g−1. More important are the high specific capacitance and the excellent reversibility and cycling stability of these electrode materials at a high current density of 10 A g−1; there was no obvious capacitance fading even after 1000 cycles.
Experimental
Cobalt (II) chloride hexahydrate (CoCl2·6H2O), nickel (II) chloride hexahydrate (NiCl2·6H2O), and urea (35 mM) were dissolved in 100 mL DI water to form a transparent solution, in which the total concentration of Co2+ and Ni2+ is 15 mM. The Co/Ni molar ratios are 1:0; 2:1; and 0:1 in the reactants, respectively (see Table 1). Then, the resulting solution was sealed in a Teflon-lined stainless autoclave, heated to 100 °C and kept at that temperature for 48 h. The products were collected by centrifugation and washed with DI water three times and with anhydrous ethanol one time, and finally dried in vacuum oven at 40 °C for 24 h. Finally, the precipitates were calcified at 300 °C for 3 h under the air atmosphere at the heating rate of 10 °C/min.| Co/Ni molar ratio in the reactants | Crystal Phase (after 300 °C calcination) | BET specific surface area [m2 g−1] | BJH pore volume [mL g−1] | Specific Capacitance [F g−1] |
|---|---|---|---|---|
| a The specific capacitance is measured at a current density of 1 A/g. b The specific capacitance is measured at a current density of 10 A/g. | ||||
| 1:0 | Co3O4 | 31.3 | 0.1536 | 60a, 31b |
| 2:1 | NiCo2O4 | 198.9 | 0.4599 | 658a, 530b |
| 1:0 | NiO | 186.4 | 0.3407 | 194a, 152b |
The morphologies were directly examined by scanning electron microscopy (SEM) using JEOL JSM-6700F at an accelerating voltage of 5 kV. EDX spectra were carried out on JEOL JSM-6300F to determine the Co/Ni molar ratio for single particle in the products via surface mapping, and collected from five different areas for every sample. Transmission electron microscopy (TEM) observations were carried out on a JEOL 2010 microscope operating both at 200 kV. X-ray diffraction (XRD) was performed on a Philips PW-1830 X-ray diffractometer with Cu kα irradiation (λ = 1.5406 Å). The step size and scan rate are set as 0.05° and 0.025°/s, respectively. X-ray fluorescence spectrometer (XRF) was carried out on JEOL JSX-3201Z to determine the Co/Ni molar ratio. Brunauer-Emmett-Teller (BET) surface areas were measured using a Coulter SA 3100 surface area analyzer.
The working electrode was prepared with the active material, acetylene black and polyvinylidene fluoride (PVDF) binder in a weight ratio of 80:10:10. The slurry was coated on graphite paper and dried under a vacuum at 100 °C at least for 12 h at an active mass loading of at least 1 mg cm−2. The electrolyte used was a 1.0 M KOH aqueous solution. The capacitive performance of the samples was evaluated on a CHI 660C electrochemical workstation using cyclic voltammetry and chronopotentiometry test with a three-electrode cell where Pt foil serves as the counter electrode and Ag/AgCl (saturated KCl solution) as the reference electrode.
Results and discussion
In the present work, the precursor compounds are metal carbonate hydroxide salts, which were formed by reacting metal cations (Co2+ and Ni2+) with CO32− and OH− anions slowly released from hydrolysis of urea in an aqueous solution. After the low temperature calcinations (300 °C), metal carbonate hydroxide salts were decomposed into porous metal oxides accompanied by the release of CO2 and H2O gases. The relevant chemical reactions involved can be represented here as eqns (1)–(3):| (Co2+,Ni2+) + CO32− + OH− → (Co, Ni)2CO3(OH)2 | (1) |
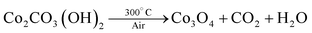 | (2) |
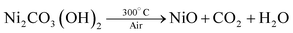 | (3) |
Co(CO3)0.5(OH)·0.11H2O (JCPDS 48-0083) and Ni2CO3(OH)2·2H2O (JCPDS 29-0868) were formed at the molar ratios of Co/Ni are 1:0 and 0:1, respectively (Fig. 1A). After 300 °C calcination, they were transformed into Co3O4 (JCPDS 65-3103) and NiO (JCPDS 47-1049), respectively, as shown in Fig. 1B. The cobalt-nickel bimetallic carbonate hydroxide salts (Ni2/3Co4/3(CO3)(OH)2) were formed at the molar ratio of Co/Ni at 2:1, and decomposed into the NiCo2O4 phase (JCPDS 20-0781) after calcination. The XRD results establish that the simple thermal decomposition method is suitable for preparing many different transition metal oxide nanomaterials, especially for multiple transition metal oxides, in which the molar ratios of transition metal elements can be easily adjusted.
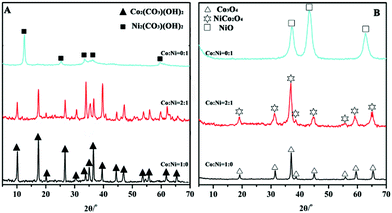 | ||
| Fig. 1 XRD patterns of (A) metal carbonate hydroxide salts (Co/Ni = 1:0, 2:1, and 0:1) and (B) the corresponding metal oxides after 300 °C calcination for 3 h. | ||
To observe the morphologies of the nanomaterials, we turned to SEM, and the resulting images of the precursors and decomposition products are shown in Fig. 2. First, the Co(CO3)0.5(OH)·0.11H2O precursor (Co/Ni = 1:0) can be figured by crystalline needles (Fig. 2A-I, II). Second, the precursor of Co-Ni bimetallic carbonate hydroxide salts obtained at the Co/Ni molar ratio of 2:1 exclusively holds a sea urchin-like morphology comprising the building blocks of needle-like nanorods (Fig. 2B-I, II). Finally, in the absence of Co2+ cations, the precursor contains only the nanoplate-based flower-like Ni2CO3(OH)2·2H2O spheres (Fig. 2C-I, II). After calcination, the overall morphologies were nearly conserved during the phase transformation from the metal carbonate hydroxide salts to the corresponding metal oxides (Fig. 2A, B, C-III). Interestingly, the precursors in the main have smooth surfaces, but the surface of the metal oxides after thermal decomposition is generally rough. For example, the cobalt carbonate hydroxide nanorods appear smooth on their surfaces (Fig. 2A-II), whereas the thermal decomposition product cobalt oxide nanorods seem to be composed of small nanoparticles (Fig. 2A-III). Overall, the morphologies are conserved during the thermal decomposition processes although the internal nanoscale structures have been changed by opening pore channels thanks to the expulsion of small gas molecules.
 | ||
| Fig. 2 SEM images of metal carbonate hydroxide salts formed at various Co/Ni molar ratios: (A-I, II) Co/Ni = 1:0; (B-I, II) Co/Ni = 2:1; (C-I, II) Co/Ni = 0:1, and the corresponding metal oxides after 300 °C calcination for 3 h: (A-III) Co3O4 (Co/Ni = 1:0); (B-III) NiCo2O4 (Co/Ni = 2:1); (C-III) NiO (Co/Ni = 0:1). | ||
We further fell back on TEM to analyze the micro- and nano-structures of the metal carbonate hydroxides. Fig. 3 shows TEM images of the metal carbonate hydroxide salts. Indeed, the cobalt carbonate hydroxide salts are in a needle-like morphology growing along the [100] direction (Fig. 3A-I, II, III). The diameter of the sharp tips is over 100 nm (Fig. 3A-II). After doping Ni atom into the cobalt carbonate hydroxide salts, the formed Co-Ni bimetallic carbonate hydroxide salts take a sea urchin-like morphology (Fig. 3B-I). The building blocks are needle-like nanorods (Fig. 3B-II). The nanorod is all single crystalline growing along the a axis direction (Fig. 3B-III). As for the nickel carbonate hydroxide products, the SEM and TEM images (Fig. 2C-I, II and Fig. SI-1, ESI†) combined reveal flower-like microspheres consisting of nanoplates. The nanoplates show a well-defined, spotty SAED pattern (inset of Fig. SI-1B, ESI†), which is consistent with a single crystal structure with the nanoplate plane being perpendicular to the [100] direction (Fig. SI-1B, ESI†).
 | ||
| Fig. 3 TEM and HRTEM images of metal carbonate hydroxide salts formed at various Co/Ni molar ratios: (A-I, II, III) Co/Ni = 1:0, (B-I, II, III) Co/Ni = 2:1(Inset: the corresponding SAED pattern). | ||
To better understand the reaction mechanisms responsible for the formation of the nanomaterials, we surveyed the product morphology as a function of reaction time. In all these experiments, urea was slowly hydrolyzed to form CO32− and OH− anions in an aqueous solution, which would react with Co2+ and Ni2+ cations initiating nucleation. We first look at the monometallic case. After the hydrolysis of urea for 1.5 h, flower like nickel carbonate hydroxide salts have formed from nanoparticles with a diameter of <100 nm (Fig. SI-2, ESI†), and cobalt carbonate hydroxide nanorods have precipitated (Fig. SI-3, ESI†). For the bimetallic case, the reactant solution remained transparent after 1.5 h of reaction due to the lower individual Co2+ and Ni2+ concentrations. Two hours later, monometallic nickel carbonate hydroxides were firstly precipitated as can be seen from the XRD pattern in Fig. 4F. The corresponding SEM images in Fig. 4A, B further reveal that nanoparticles with a diameter of <100 nm were initially formed and then transformed into a plate-shape structure. This process in the presence of both Co2+ and Ni2+ is in much the same way as the formation of nickel carbonate hydroxides in the presence of only the monometallic nickel reactants (Fig. SI-2, ESI†). On prolonging the reaction time to 2.5 h, the monometallic nickel carbonate hydroxide nanoplates were changed into the flower-like spheres, which started to serve as a substrate for growing bimetallic (Ni, Co) carbonate hydroxide nanorods by nanoscale dissolution-recrystallization (Fig. 4C, D, F and Fig. SI-4, ESI†). A further increase of the reaction time to 3.0 h led to a complete transformation of the flower-like nickel carbonate hydroxide into the sea urchin-like bimetallic (Ni, Co) carbonate hydroxide salts (Fig. 4E and 4F).
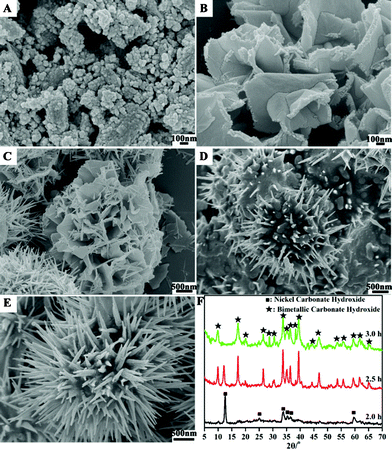 | ||
| Fig. 4 SEM images of metal carbonate hydroxide salt formed at the Co/Ni molar ratio of 2:1 at different reaction stages: (A, B) 2.0 h; (C, D) 2.5 h; (E) 3.0 h; and (F) the corresponding XRD pattern. | ||
Classically, the homogeneous nucleation rate for a spherical nucleus (JN) is given by JN = Aexp(−ΔG*N/kT), ΔG*N = −16πσ3ν2/3(kTlnSR)2 Where A is a pre-exponential factor; ΔG* corresponds to the nucleation activation energy; k is the Boltzmann constant; T is the temperature; σ is the interfacial free energy per unit surface area; ν is the molecular volume; SR corresponds to the relative supersaturation. Considering that the solubility product constants (Ksp) at 25 °C of CoCO3 (8.0 × 10−13) and Co(OH)2 (2.5 × 10−16) are smaller than those of NiCO3 (6.6 × 10−9) and Ni(OH)2 (2.8 × 10−16), a lower supersaturation for nickel carbonate hydroxide than that for cobalt carbonate hydroxide is expected, running counter to the first nucleation of the nickel carbonate hydroxide. However, this could be reversed if the nickel carbonate hydroxide has a significantly lower interfacial energy than that of cobalt carbonate hydroxide. This seems to be supported by the initial preferential formation of the nickel carbonate hydroxide nanoplates bounded mainly by the low-energy {100} surfaces. The tendency to form nanoplates may be related to the intrinsic square coordination structure of Ni2+ (d8).
The formation of the flower-like nickel carbonate hydroxide appears to have facilitated the subsequent nucleation and growth of the more thermodynamically stable bimetallic (Ni, Co) carbonate hydroxide phase in nanorod morphology by localized dissolution-reprecipitation. More specifically, the monometallic nickel carbonate hydroxide flowers served as substrates for the growth of the bimetallic carbonate hydroxide nanorods. The bimetallic carbonate hydroxide shares a similar crystal structure with the cobalt carbonate hydroxide, as can be seen from the XRD patterns in Fig. 1. Here the transformation from the monometallic Ni carbonate hydroxide nanoplates to the bimetallic (Ni, Co) carbonate hydroxide can only be mediated by the solvent through perhaps a localized dissolution-reprecipitation process. Indeed, we have observed a gradual disappearance of the nickel carbonate hydroxide nanoplates concomitant with the formation of the bimetallic (Ni, Co) carbonate hydroxide nanorods (see Fig. 4 and Fig. SI-4, ESI†). Of note, the similar lattice constants between the a-axis of Ni2CO3(OH)2·2H2O (orthorhombic, 10.18 × 27.4 × 3.22 Å) and the b-axis of Co(CO3)0.5(OH)·0.11H2O (orthorhombic, 8.79 × 10.15 × 4.433 Å) may have helped the nucleation and growth of the bimetallic carbonate hydroxide. Because of the different electronic structure of Co2+ (prefer octahedral coordination) from that of Ni2+ (prefer square-planar coordination), the growth along the c-axis is the fastest now, leading to the formation of the bimetallic carbonate hydroxide nanorods. The growth of the nanorods was accompanied by the depletion of the reactant concentrations, which can explain the tapering of the growing nanorods and the final formation of the sea urchin-like bimetallic (Ni, Co) carbonate hydroxide.16
After calcination at 300 °C for 3 h, the metal carbonate hydroxide salts were decomposed into the corresponding metal oxides, as seen from Fig. 1. The metal oxide products such as Co3O4 nanorods (Fig. 5A-I), sea urchin-like Co-Ni bimetal oxides (Fig. 5B-I) and nanoplate-based flower-like NiO spheres (Fig. 5C-I) have inherited the overall morphologies of the corresponding metal carbonate hydroxides salts, in agreement with the SEM results (Fig. 2). As expected, the observed difference is that the thermally transformed products are polycrystalline and porous (Fig. 5A, B, C -II) clearly arising from the escape of gas molecules of CO2 and H2O. Such an hierarchical structure with interconnected pores is a crucial feature to function as an electroactive material thanks to the facilitated electrolyte and electron transport.17 Further pore structural characterizations will be presented below.
 | ||
| Fig. 5 TEM images of metal oxides: (A-I, II) Co3O4 nanorods, (B-I, II) sea urchin-like NiCo2O4 spheres, and (C-I, II) nanoplate-based flower-like NiO spheres (Inset: the corresponding SAED pattern). | ||
The specific surface areas and pore volumes are important parameters closely related to the specific capacitances of electroactive materials. The porous characteristics of the products were investigated via Brunauer-Emmett-Teller (BET) gas-sorption measurements. The specific surface areas, pore volumes, and the corresponding Barrett-Joyner-Halenda (BJH) pore size distribution plots were calculated from N2 adsorption-desorption isotherms of the products, and are presented in Fig. 6 and Table 1. All of the adsorption isotherms can be categorized as type IV (Fig. 6A).18 The hysteresis loop in the Co3O4 samples can be categorized as Type H1, revealing a narrow distribution of pore size (average pore diameter: 16.5 nm) as shown in Fig. 6B. The distinct hysteresis loop for the NiCo2O4 samples shifts to 0.6–0.8 P/P° compared to 0.8–1.0 P/P° for the Co3O4 samples, indicating a smaller mean pore size (average pore diameter: 5 nm, Fig. 6B). The hysteresis loop for the NiO samples should belong to Type H3, featuring slit-shaped pores (Fig. 6A). The specific surface areas of the metal oxide products Co3O4, NiCo2O4, and NiO have been estimated at 31.3, 198.9, and 186.4 m2 g−1, and charted in Table 1. The BET results are in good agreement with the TEM observations discussed above, outfitting the structural characteristics of the products needed for their electrochemical studies below.12
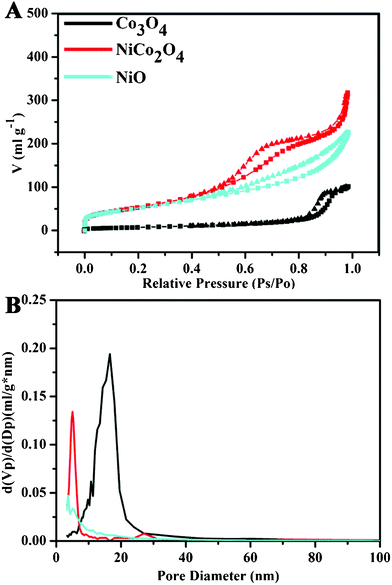 | ||
| Fig. 6 (A) Nitrogen adsorption (■) and desorption (▾) isotherms of Co3O4, NiCo2O4 and NiO measured at standard temperature and pressure, and (B) the corresponding BJH pore size distribution plots. | ||
The specific capacitances are determined by cyclic voltammetry (CV) and galvanostatic charge-discharge measurement. Fig. 7 shows CV curves of Co3O4, NiCo2O4, and NiO electrodes, as measured in 1.0 M KOH solution. The voltage was swept from −0.1 to 0.5 V at a scanning rate of 5 mV s−1. The distinct pairs of redox peaks are clearly observed in the anodic and cathodic sweeps, sitting on a broad background. Clearly, the redox peaks deliver the faradic pseudocapacity based on the surface redox reactions of Ni2+ to Ni3+ and Co2+ to Co3+. Co3O4 and NiO electrodes show an anodic peak at around 0.40 and 0.36 V, and a cathodic peak at about 0.35 and 0.28 V, respectively, associated with the redox couples of Co3O4/CoOOH and NiO/NiOOH.7,19 The CV curve of NiCo2O4 electrode shows a more widely separated pair of redox peaks and encloses a much larger area than those of the NiO and Co3O4 electrodes. In fact, the peaks at around 0.39 V and 0.27 V mainly correspond respectively to the oxidation of Co2+ and the reduction of Ni3+, as previously reported by Hu et al.,14 while the peaks of Ni2+ oxidation and the Co3+ reduction are not discernible, but can at times be observed (Fig. SI-5, ESI†), dependant on the electrode preparation. Therefore, although there is a large separation (120 mV) between the major anodic and cathodic peaks, the actual peak separation is only 40 mV for the (Co2+, Co3+) pair and 70 mV for the (Ni2+, Ni3+) pair.
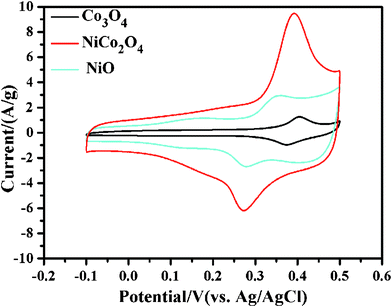 | ||
| Fig. 7 CV curves of Co3O4, NiCo2O4, and NiO electrodes sweeping from −0.1 V to 0.5 V at the potential scanning rate of 5 mV s−1 in 1.0 M KOH solution. | ||
To further quantify the specific capacitances, galvanostatic charge-discharge curves were measured in the same working cells as used in the CV measurements (see Fig. 8A). The specific capacitances calculated from the corresponding charge-discharge curves are listed in Table 1. The specific capacitance of NiCo2O4 electrode is obviously much higher than those of the single component electrodes, i.e., the Co3O4 and NiO electrodes. This can be understood by the richer redox reactions (contributed by both Co2+ and Ni2+ ions) and much higher electrical conductivity of NiCo2O4 materials than those of the monometallic oxide materials.9 The energy efficiencies of Co3O4, NiCo2O4, and NiO electrodes are, respectively, 57.5%, 70.8%, and 63.2% in the initial cycles, and increase to 64.0%, 73.6%, and 69.2% after 100 cycles, as calculated from the galvanostatic charge-discharge curves (I = 10 A/g). The good electrochemical performance of NiCo2O4 electrode was further confirmed by the electrochemical impedance spectroscopy (EIS) measurements. Fig. 9 shows the Nyquist plots of the EIS spectra of Co3O4, NiCo2O4, and NiO electrodes, respectively. The EIS data were fitted by an equivalent circuit consisting of a bulk solution resistance Rs, a charge-transfer Rct, a pseudocapacitive element Cp, and a constant phase element (CPE), as shown in the inset of Fig. 9. The bulk solution resistance Rs and charge-transfer resistance Rct can be estimated from the intercepts of the high frequency semicircle with the real axis at Rs and (Rs+Rct), respectively.20 From the data, the solution resistance Rs of Co3O4, NiCo2O4, and NiO electrodes is 4.7, 5.3, and 4.4 Ω, respectively, while the charge-transfer resistance Rct is 5.2, 0.9, and 4.0 Ω in the same order. This clearly demonstrates that the NiCo2O4 electrode has a much better charge-transfer property against the Co3O4 and NiO electrodes. Notice that the charge-transfer resistance Rct, also called Faraday resistance, is a limiting factor for the specific power of a supercapacitor,2,21 hence the low Faraday resistance of our NiCo2O4 electrode really means a high specific power achievable.
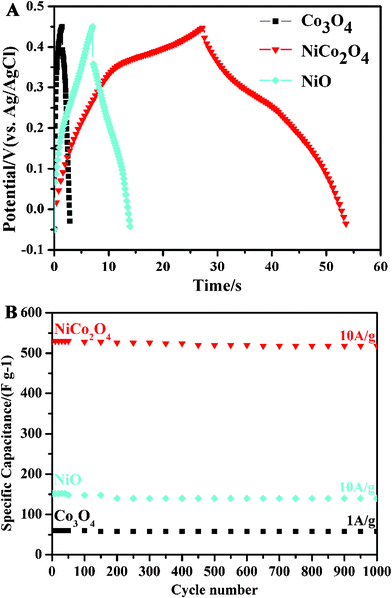 | ||
| Fig. 8 (A) Galvanostatic charge-discharge curves of Co3O4, NiCo2O4, and NiO electrodes at a charge-discharge current density of 10 A g−1, and (B) the corresponding stability test. | ||
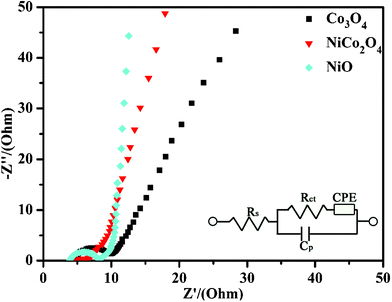 | ||
| Fig. 9 Nyquist plot of the EIS of the Co3O4, NiCo2O4, and NiO electrodes (Inset: the equivalent circuit diagram of different elements from the EIS analysis). | ||
Finally, cycle stabilities of the electrodes have been demonstrated as shown in Fig. 8B. After 1000 cycles of charge-discharge, the specific capacitances degraded by 3.33% and 7.89%, respectively, for the Co3O4 electrode at a current density of 1 A g−1 and for the NiO electrode at a current density of 10 A g−1. The loss of the specific capacitance is likely related to the degradation of the active material microstructure in the process of OH−insertion (extraction) during oxidation (reduction).7 The relevant redox reactions of Ni2+ to Ni3+ and Co2+ to Co3+ that occur at the surfaces of electroactive materials can be expressed by the following equations:7,22
 | (4) |
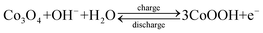 | (5) |
For the NiCo2O4 electrode, however, the specific capacitances suffered only a loss of 1.57%, at a current density of 10 A g−1. Clearly, the excellent cycling stability of these nickel cobaltite spinel electrodes can be attributed to the well-connected hierarchical porous structure and the good electrical conductivity, which ensure fast transport of electrolyte ions and electrons at a high current density.14,23
The supercapacitor performance of our nanomaterials is to be compared with that of the relevant materials. Zhang et al.7 reported that the specific capacitance of hierarchical NiO microspheres was found to decrease from 710 to 525 F g−1 as the current density was increased from 1 to 4 A g−1, even when 20 wt.% carbon black was used for preparing the electrodes. Co3O4 nanostructures are normally reported to have low specific capacitance (less than 100 F g−1).4 Recently, Wang et al.24 prepared mesoporous Co3O4 using mesoporous silica as hard templates and obtained a specific capacitance of 370 F g−1, but 30 wt.% carbon black had to be used in preparing the electrodes. As mentioned above, a NiCo2O4 and Ni(OH)2 mixed aerogel prepared by a relatively complex epoxide-driven sol-gel-supercritical process could achieve a specific capacitance of 634 F g−1, but suffered a significant decrease to 420 F g−1 after 300 °C calcination.14 In our case, the nickel cobaltite spinel (NiCo2O4) nanomaterials have achieved specific capacitances of 658 F g−1 at a current density of 1 A g−1 when only 10 wt.% carbon black was used in preparing the electrodes. Even at a high charge-discharge current density (10 A g−1), the specific capacitances could preserve 530 F g−1, and only had 1.57% capacitance loss after 1000 cycles. Thus, the sea urchin like porous nickel cobaltite spinel nanomaterials really stand out in terms of their high specific capacitance, high rate capability, and excellent cycling stability.
A good performing pseudo-capacitor entails fast and reversible redox reactions between the electrolyte and electroactive species on the electrode surface. Thus, the number of electroactive sites of electrode surface and the transport rates of electrolyte ions and electrons in Faradic reactions are important parameters for the faradic reactions, which are respectively influenced by the specific surface areas and the pore structure and electric conductivity. Prior preparations of nickel cobaltite spinel materials mostly resulted in low specific surface areas. For example, nickel-cobalt carbonate hydroxide precursors from direct co-precipitation, when thermally decomposed, gave rise to nickel cobaltite spinel with only a specific surface area of <90 m2 g−1.15 Even with a more complex epoxide-driven sol-gel process followed by drying in supercritical carbon dioxide, the resulting NiCo2O4 and Ni(OH)2 mixed aerogel had a specific surface area of only ∼120 m2 g−1, which was dramatically decreased to 43 m2 g−1 after calcination at 300 °C.14 In contrast, due to the controlled formation of precursors, our nickel cobaltite spinel nanomaterials are stable and support a high specific surface area of 198.9 m2 g−1, which can provide profuse electroactive surface sites for faradic reactions.
In addition, the nickel cobaltite spinel nanomaterials produced by our method have a hierarchical pore structure which can facilitate fast electrolyte and electron transport, a feature especially important for high rate electrochemical capacitors. It is well documented that macropores can serve as “ion buffering reservoirs”, mesopores are capable of overcoming the primary kinetic limits of electrochemical processes, and micropores can enhance the charge storage.25 To our delight, our nickel cobaltite spinel nanomaterials are imbued with such a hierarchical pore structure with interconnected pores at macro-, and meso-scales, as can be seen in the TEM images (Fig. 5) and BET gas-sorption results (Fig. 6). On top of that, the nickel cobaltite spinel nanomaterials have at least two orders of magnitude higher electric conductivity than those of nickel oxides and cobalt oxides separately, which is essential for fast electron transport, especially at high current densities. In total, the nickel cobaltite spinel nanomaterials developed here have the structural and electrical characteristics suited for pseudocapacitors.
Conclusions
In sum, we have established that a sea urchin-like bimetallic (Ni, Co) carbonate hydroxide precursor was formed through a sequential crystallization process. The precursor was then thermally decomposed at a low temperature into porous NiCo2O4 spinel with the sea urchin morphology largely conserved. During the precursor formation, nickel carbonate hydroxide rather than bimetallic (Ni, Co) carbonate hydroxide was firstly nucleated, and crystallized into the flower-like microspheres. Then, at the edge of the nanoplates in the flower-like microspheres, bimetallic (Ni, Co) carbonate hydroxide was heterogeneously nucleated through a nanoscale dissolution-recrystallization process, and anisotropically grown into nanorods. Finally, a sea urchin-like morphology was obtained. After low temperature calcination (300 °C), porous NiCo2O4 spinel was formed with a largely conserved sea urchin-like morphology. This method, in comparison with the thermal decomposition of directly co-precipitated precursors, can achieve high specific surface areas. Moreover, it is simpler and more effective than the sacrificial template methods for preparing stable nanostructures with hierarchical pores including macro-, and meso-pores, suitable for fast electrolyte transport in high-capacity and high-rate electrochemical devices. Indeed, these novel NiCo2O4 nanomaterials have demonstrated high specific capacitances of 658 F g−1 at a current density of 1 A g−1, significantly higher than those of the porous Co3O4 (60 F g−1) and NiO (194 F g−1) electrodes. Especially, at a high charge-discharge current density (10 A g−1), the specific capacitance can preserve high values at 530 F g−1, and the capacitance decay was insignificant even after 1000 cycles of charge-discharge, revealing the excellent stability of the nanomaterials. As a whole, this work demonstrates the promise of nanostructuring next-generation supercapacitor materials by controllably preparing a low-cost binary oxide hierarchical porous nanostructurevia a simple precursor decomposition process.Acknowledgements
This work was supported by the Research Grants Council of Hong Kong under the JRF No. 604809.References
- (a) J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS; (b) P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors, Kulwar Academic/Plenum Publishers, New York, 1999. Search PubMed.
- C. C. Hu, W. C. Chen and K. H. Chang, J. Electrochem. Soc., 2004, 151, A281 CrossRef CAS.
- (a) S. L. Xiong, C. Z. Yuan, M. F. Zhang, B. J. Xi and Y. T. Qian, Chem.–Eur. J., 2009, 15, 5320 CrossRef CAS; (b) T. Zhu, J. S. Chen and X. W. Lou, J. Mater. Chem., 2010, 20, 7015 RSC.
- Y. Y. Gao, S. L. Chen, D. X. Cao, G. L. Wang and J. L. Yin, J. Power Sources, 2010, 195, 1757 CrossRef CAS.
- (a) H. Inoue, Y. Namba and E. Higuchi, J. Power Sources, 2010, 195, 6239 CrossRef CAS; (b) X. J. Zhang, W. H. Shi, J. X. Zhu, W. Y. Zhao, J. Ma, S. Mhaisalkar, T. L. Maria, Y. H. Yang, H. Zhang, H. H. Hng and Q. Y. Yan, Nano Res., 2010, 3, 643 CrossRef CAS; (c) Y. Z. Zheng, H. Y. Ding and M. L. Zhang, Mater. Res. Bull., 2009, 44, 403 CrossRef CAS; (d) X. M. Liu, X. G. Zhang and S. Y. Fu, Mater. Res. Bull., 2006, 41, 620 CrossRef CAS; (e) Y. G. Wang and Y. Y. Xia, Electrochim. Acta, 2006, 51, 3223 CrossRef CAS; (f) M. Q. Wu, J. H. Gao, S. R. Zhang and A. Chen, J. Porous Mater., 2006, 13, 407 CrossRef CAS.
- C. Z. Yuan, X. G. Zhang, L. H. Su, B. Gao and L. F. Shen, J. Mater. Chem., 2009, 19, 5772 RSC.
- (a) Z. P. Feng, G. R. Li, J. H. Zhong, Z. L. Wang, Y. N. Ou and Y. X. Tong, Electrochem. Commun., 2009, 11, 706 CrossRef CAS; (b) H. S. Nam, J. K. Yoon, J. M. Ko and J. D. Kim, Mater. Chem. Phys., 2010, 123, 331 CrossRef CAS; (c) H. S. Nam, J. S. Kwon, K. M. Kim, J. M. Ko and J. D. Kim, Electrochim. Acta, 2010, 55, 7443 CrossRef CAS; (d) X. H. Tang, H. J. Li, Z. H. Liu, Z. P. Yang and Z. L. Wang, J. Power Sources, 2010, 196, 855 CrossRef; (e) Y. Lei, B. Daffos, P. L. Taberna, P. Simon and F. Favier, Electrochim. Acta, 2010, 55, 7454 CrossRef CAS.
- M. R. Tarasevich and B. N. Efremov, Electrodes of Conductive Metallic oxides Part A, Elsevier, USA, 1982 Search PubMed.
- (a) D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS; (b) D. Gu, H. Bongard, Y. H. Deng, D. Feng, Z. X. Wu, Y. Fang, J. J. Mao, B. Tu, F. Schuth and D. Y. Zhao, Adv. Mater., 2010, 22, 833 CrossRef CAS; (c) Z. R. Tian, W. Tong, J. Y. Wang, N. G. Duan, V. V. Krishnan and S. L. Suib, Science, 1997, 276, 926 CrossRef CAS; (d) B. Smarsly, D. Grosso, T. Brezesinski, N. Pinna, C. Boissiere, M. Antonietti and C. Sanchez, Chem. Mater., 2004, 16, 2948 CrossRef CAS.
- (a) Z. X. Wu, Q. A. Li, D. Peng, P. A. Webley and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 12042 CrossRef CAS; (b) S. M. Zhu, Z. Y. Zhou, D. Zhang and H. H. Wang, Microporous Mesoporous Mater., 2006, 95, 257 CrossRef CAS.
- J. Roggenbuck and M. Tiemann, J. Am. Chem. Soc., 2005, 127, 1096 CrossRef CAS.
- J. Lee, M. C. Orilall, S. C. Warren, M. Kamperman, F. J. Disalvo and U. Wiesner, Nat. Mater., 2008, 7, 222 CrossRef CAS.
- T. Y. Wei, C. H. Chen, H. C. Chien, S. Y. Lu and C. C. Hu, Adv. Mater., 2010, 22, 347 CrossRef CAS.
- D. G. Klissurski and E. L. Uzunova, Chem. Mater., 1991, 3, 1060 CrossRef CAS.
- X. P. Shen, J. Q. Sun, G. X. Wang, J. Park and K. M. Chen, Mater. Res. Bull., 2010, 45, 766 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- T. Y. Wei, C. H. Chen, K. H. Chang, S. Y. Lu and C. C. Hu, Chem. Mater., 2009, 21, 3228 CrossRef CAS.
- (a) K. P. Wang and H. S. Teng, J. Electrochem. Soc., 2007, 154, A993 CrossRef CAS; (b) C. W. Huang and H. S. Teng, J. Electrochem. Soc., 2008, 155, A739 CrossRef CAS.
- (a) J. F. Zang, S. J. Bao, C. M. Li, H. J. Bian, X. Q. Cui, Q. L. Bao, C. Q. Sun, J. Guo and K. R. Lian, J. Phys. Chem. C, 2008, 112, 14843 CrossRef CAS; (b) L. H. Bao, J. F. Zang and X. D. Li, Nano Lett., 2011, 11, 1215 CrossRef CAS.
- (a) V. Srinivasan and J. W. Weidner, J. Power Sources, 2002, 108, 15 CrossRef CAS; (b) Y. Y. Gao, S. L. Chen, D. X. Cao, G. L. Wang and J. L. Yin, J. Power Sources, 195, 1757 CrossRef CAS.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878 CrossRef CAS.
- G. X. Wang, H. Liu, J. Horvat, B. Wang, S. Z. Qiao, J. Park and H. Ahn, Chem.–Eur. J., 2010, 16, 11020 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images and SAED pattern of nickel carbonate nanoplate; SEM images of nickel or cobalt carbonate hydroxide after 1.5 h reaction; and SEM images of bimetallic (Ni, Co) carbonate hydroxide nucleated on the edge of nickel carbonate hydroxide nanoplate. See DOI: 10.1039/c1ra00342a |
| This journal is © The Royal Society of Chemistry 2011 |