CO2 chemistry: task-specific ionic liquids for CO2 capture/activation and subsequent conversion
Zhen-Zhen
Yang
,
Ya-Nan
Zhao
and
Liang-Nian
He
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: heln@nankai.edu.cn; Fax: (+86)-22-2350-3627; Tel: (+86)-22-2350-1493
First published on 22nd August 2011
Abstract
Ionic liquids (ILs), a kind of novel green medium composed entirely of cations and anions, have attracted considerable attention due to their unique properties such as non-volatility, tunable polarity, high stability and so on. In this article, the latest progress on the absorption and subsequent conversion of CO2 by using ILs as absorbents, catalysts or promoters will be summarized. The chemical absorption performance of ILs, especially task-specific ionic liquids (TSILs) such as amino-functionalized ILs, superbase-derived protic ILs has been systematically illustrated. Although significant advances have been made, extensive energy input in the desorption process to recover absorbents would still be a crucial barrier to realizing practical carbon capture and sequestration (CCS). On the other hand, efficient applications of CO2 in the synthesis of valuable compounds such as organic carbonates, urea derivatives, oxazolidinones and formic acid can also be promoted by employing TSILs as catalysts/reaction media. We anticipate that an integration of chemical capture of CO2 with its utilization, a so-called CO2 capture and utilization (CCU) protocol would be an ideal strategy to solve the energy penalty problem in common CCS without the need for additional heat desorption. The essence of this CCU concept is to use TSILs for CO2 capture and substantial activation, which could allow catalytic transformation of CO2 to be accomplished smoothly under low pressure (ideally at 1 atm).
 Zhen-Zhen Yang | Zhen-Zhen Yang was born in 1986 in Henan Province, P. R. China. She studied chemistry at Central China Normal University and now in the first grade of the doctor program at the State Key Laboratory of Elemento-Organic Chemistry, Nankai University, under the supervision of Prof. Liang-Nian He. Her research interests cover green synthetic chemistry and CO2 chemistry (CO2 capture, CO2 activation and chemical transformation into fuels/value-added chemicals). |
 Ya-Nan Zhao | Ya-Nan Zhao was born in 1988 in Heibei province, P. R. China. She studied chemistry at Hebei Normal University and now she is studying organic chemistry at the State Key Laboratory of Elemento-Organic Chemistry in Nankai University, under the supervision of Prof. Liang-Nian He. She will receive her master's degree in 2013. Her research interests include CO2 capture and chemical conversion utilizing task-specific ionic liquids as absorbents, catalysts or promoters. |
 Liang-Nian He | Professor Liang-Nian He received his Ph.D. degree from Nankai University in 1996 under the guidance of academician Ru-Yu Chen. He then worked as a Chinese postdoctoral fellow with academician Ren-Xi Zhuo at Wuhan University. He had worked as a Postdoctoral Research Associate at the National Institute of Advanced Science and Technology, Japan from 1999 to 2003 before joining Nankai University in April 2003. He has over 120 scientific publications and 6 patents. He also edited 6 books and chapters, delivered more than 30 invited lectures at international/national conferences and universities and research organizations. Current research interests cover green chemistry, CO2 chemistry, catalysis in green solvents and biomass conversion (e.g. castor-related energy). |
Introduction
Carbon dioxide from combustion of fossil fuels (coal, petroleum and natural gas) is regarded as the most significant greenhouse gas; hence, CO2 chemistry (capture and conversion)1–6 has attracted significant attention from the scientific community thanks to global warming. CO2 capture from flue gases in industrial combustion processes is now being considered as a serious option to reduce or mitigate the so-called green-house effect.1 On the other hand, removal of CO2 from natural gas is of vital importance to maintain and expand the availability of these clean-burning, efficient fuel sources.3 The conventional technology for the capture of CO2 has been applied in industry through chemical adsorption by using an aqueous solution of amine, which has some advantages, such as low cost, high absorption efficiency and capacity.7,8 However, the use of aqueous amine solutions may also pose serious inherent drawbacks, including loss of solvent, corrosion of facility, and high energy demand for regeneration of the absorbent.8,9 Therefore, alternative absorbents that could facilitate separation of CO2 from gas mixtures, e.g. flue gas, natural gas/syngas, without concurrent loss of the solvent into the gas stream would be these liquids, which have negligible volatility, making them be highly desired. ILs, especially TSILs, a kind of novel media consisting of functionalized ions, could result in a non-contaminated target gas especially attractive for the absorption of gases, showing great potential as an alternative for such applications.1,3,10–12On the other hand, as an abundant, nontoxic, non-flammable, easily available, and renewable carbon resource, CO2 is very attractive as an environmentally friendly feedstock for making commodity chemicals, fuels, and materials.4,13–18 Although CO2 utilization is unlikely to consume significant quantities of CO2, it can be a significant strategy for the development of sustainable and safe processes. Development of catalytic processes for chemical transformation of CO2 into useful compounds is of paramount importance from a standpoint of C1 chemistry and green chemistry. However, few industrial processes utilize CO2 as a raw material, because CO2 is the most oxidized state of carbon, namely CO2 could be a thermodynamically stable molecule. In this context, the biggest obstacle for establishing industrial processes for CO2 conversion would be due to its low energy level.14 In short, its inherent thermodynamic stability and kinetic inertness hinder the development of efficient catalysts that achieve CO2 activation and subsequent its functionalization. Accordingly, only if we understand the underlying principles of CO2 activation, can the goal of using CO2 as an environmentally friendly and economically feasible source of carbon be achieved.
Great progress in catalytic utilization of CO2 as an alternative to phosgene and/or carbon monoxide in organic synthesis has been seen. CO2 has been found to be a useful synthon to react with small-membered ring compounds,19–21 unsaturated compounds,22 hydrogen,23 amines24,25 and so on for constructing C–C, C–O and C–N bond on the basis of CO2 activation through molecular catalysis. ILs are composed of cation/anion (Lewis acid and Lewis base sites) combinations, which may be able to activate CO2 molecules and simultaneously interact with the other substrates, especially TSILs with various functionalized groups.26 For example, in the cycloaddition reaction of CO2 with epoxides, besides activation of CO2, hydroxyl-functionalized or zinc halides-contained ILs could also coordinate with the oxygen atom of epoxides, thus resulting in epoxide's activation.27,28 We have also developed Lewis basic ILs to catalyze CO2 conversion. This kind of functionalized ILs contain tertiary nitrogen in the cation, and thus have the potential reacting with CO2 to form carbamate species, which can be considered as an activated form of CO2.20,21 Indeed, ILs have been found wide applications in terms of CO2 conversion.
In the past decades, numerous strategies have been proposed for chemical absorption of CO2. Although significant advances have been made, there are still inherent drawbacks such as extensive energy consumption for CO2 desorption, low capture efficiency and slow sorption kinetics to be addressed.1,3,10–12 In particular, extensive energy input in desorption process would be a crucial barrier to realize practical CCS. Hence, reducing the energy requirement is an essential prerequisite for a breakthrough in absorption techniques. Moreover, the reactions involving CO2 are commonly carried out at high pressure, which may not be economically suitable and also pose safety concerns. The challenge is to develop efficient catalysts that are capable of activating CO2 under low pressure (preferably at 1 atm), and thus incorporating CO2 into organic molecules catalytically. In this review, the latest progress on the absorption or conversion of CO2 using ILs as absorbents, catalysts or promoters will particularly be summarized. On the other hand, those approaches in ILs such as electrochemical reduction of CO229 and microwave-assisted CO2 conversion30 could be very important strategies for CO2 utilization, but will not be discussed in detail herein. We hope this article will pave the way for an alternative approach to solve the energy penalty problem in the CCS process. In this regard, we have proposed the CCU concept as one part of CO2 chemistry.5,6 The essence is to use TSILs for CO2 capture and whereby substantial activation, which renders the reaction system suitable for accomplishing chemical transformation of CO2 under low pressure (ideally at 1 atm), getting rid of the desorption step.31–33
Carbon dioxide capture by task-specific ionic liquids
Advantages of using ionic liquids for CO2 absorption
ILs are salts composed of distinct cations and anions that are capable of facilely tuning, and whereby can be designed for task-specific applications through smart choice of the respective cation and/or anion. Nowadays, ILs have been extensively investigated with a wide range of interesting applications,30 because of their limitless attractive properties, such as wide liquid temperature ranges, good thermal stabilities, high ionic conductivity, and high solvation interactions with both polar and nonpolar compounds. On the other hand, special attention should be paid to the toxicity issue34–37 related to ILs, in particular, they are harmful to aquatic organisms.In particular, ILs have attracted significant attention in the field of gas separation from the scientific community due to their great value for chemical research and potential applications in industry.38 Because of their distinctive properties such as high thermal stability, negligible vapor pressure, high loading capacity, easy recyclability and diversiform structure/property modulation, environmentally friendly ILs have been widely used as green catalysts/solvents in a wide range of chemical reactions.26,39–42 In the past decades, design and synthesis of functionalized ILs as green absorbents in gas separation offer a new opportunity for developing novel capture systems that are capable of reversibly capturing CO2 with a high capacity and absorption rate.43–45 The negligible volatility of ILs results in a non-contaminated target gas, making them especially attractive for the absorption of gases. More importantly, TSILs can be designed according to the actual needs of industrial CO2 capture process, obtaining industrial attractive sorbent materials with high capacity and energy-saving for CO2 absorption. In 2003, ILs were used as catalysts in industrial production.46 However, further exploration is still needed for the utilization of ILs as a new generation of CO2 absorbents used in industrial decarburization. Notably, to be utilized as CO2 absorbents in industral application, ILs should possess three basic characteristics: high capacity, low viscosity (better fluidity) and economic benefits (for bulk synthesis and utilization) as well as a lower energy requirement for regeneration of the IL.
Amino-functionalized ionic liquids as CO2 sorbents
Davis and co-workers reported the first example of the chemisorption of CO2 that employed an amino-functionalized TSIL [APBIm][BF4] (Scheme 1). Those results showed that 0.5 mole CO2 can be captured per mole of IL with a gravimetric capacity of about 7.4% for 3 h under ambient pressure as shown in Scheme 1.10 The process of CO2 uptake has been repeatedly recycled five times with no observed loss of efficiency, CO2 being extruded from the IL upon heating (80–100 °C) for several hours under vacuum. In FT-IR, the spectrum of the CO2 treated material manifests a new absorption at 1666 cm−1, consistent with a carbamate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch. A new resonance observed at 158.11 ppm can be attributed to a carbamate carbonyl carbon. However, the relatively high viscosity of [APBIm][BF4] limits its eventual use in large-scale gas scrubbing applications.
O stretch. A new resonance observed at 158.11 ppm can be attributed to a carbamate carbonyl carbon. However, the relatively high viscosity of [APBIm][BF4] limits its eventual use in large-scale gas scrubbing applications.

As readily accessible chiral molecules, natural α-amino acids (AAs) have many merits, such as diverse structures, low cost, high thermal stability, bio-compatibility, easy regeneration and biodegradability. Amino acids have already been derived as both anions and cations in ILs.47 A series of phosphonium ILs with amino acids as anions [P(C4)4][AA] (Scheme 2) have been supported on porous silica gel for CO2 absorption due to their high viscosity (226.69–744.71 mPa s).48 As shown in Fig. 1, the absorption of CO2 by [P(C4)4][β-Ala]-SiO2, [P(C4)4][Gly]-SiO2 and [P(C4)4][Ala]-SiO2 can reach equilibrium in less than 100 min. One mole of NH2 can absorb half mole of CO2. For the possible reaction mechanism (Scheme 3), CO2 is attacked by the free electron pair of the N atom on the –NH2 group and thus forms a hydrogen bond with the NH2 group of another AA−.49 The absorbed CO2 can be released in a vacuum at room temperature over several hours. Four cycles of absorption/desorption can be repeated and no changes in the absorption capacity or the rates are observed.
![Cycles of CO2 absorption of [P(C4)4][AA]-SiO2. □ = [P(C4)4][Gly]-SiO2, ○ = [P(C4)4][Ala]-SiO2, and △ = [P(C4)4][β-Ala]-SiO2. Reprinted with permission from ref. 48. Copyright 2006 John Wiley & Sons.](/image/article/2011/RA/c1ra00307k/c1ra00307k-f1.gif) | ||
| Fig. 1 Cycles of CO2 absorption of [P(C4)4][AA]-SiO2. □ = [P(C4)4][Gly]-SiO2, ○ = [P(C4)4][Ala]-SiO2, and △ = [P(C4)4][β-Ala]-SiO2. Reprinted with permission from ref. 48. Copyright 2006 John Wiley & Sons. | ||
![Structures of [P(C4)4][AA] and amino acids anions.48](/image/article/2011/RA/c1ra00307k/c1ra00307k-s2.gif) | ||
| Scheme 2 Structures of [P(C4)4][AA] and amino acids anions.48 | ||
![Proposed absorption mechanism between [P(C4)4][β-Ala] and CO2.48](/image/article/2011/RA/c1ra00307k/c1ra00307k-s3.gif) | ||
| Scheme 3 Proposed absorption mechanism between [P(C4)4][β-Ala] and CO2.48 | ||
Dual amino-functionalized phosphonium ILs, (3-aminopropyl) tributylphosphonium AA salts ([aP4443][AA]) (Scheme 4) supported on SiO2 have been found to be efficient for equimolar CO2 absorption.50 The molar ratio of amino group to absorbed CO2 is still 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
![Structure of the dual amino-functionalised phosphonium ILs ([aP4443][AA]).50](/image/article/2011/RA/c1ra00307k/c1ra00307k-s4.gif) | ||
| Scheme 4 Structure of the dual amino-functionalised phosphonium ILs ([aP4443][AA]).50 | ||
As reported above, the use of [P(C4)4][AA] as liquid materials or solvents are limited due to their viscous nature. But novel AAILs using symmetric tetraalkyl ammonium ([TAA]) as cations are found to be liquid below 50 °C (Scheme 5).51 In particular, among them tetraethyl ammonium alanine ([N2222][L-Ala]) is found to have the lowest viscosity of 81 mPa s. The results of CO2 absorption into [N2222][L-Ala] and [N2222][β-Ala] at 40 °C reveal that the absorption equilibria could all be reached within 60 min with 2:1 molar ratio of AAIL/CO2 absorbed. The absorbed CO2 can be extruded from the [TAA][AA] upon heating at 50 °C for 4 h under vacuum (≈ 0.1 KPa). The recovered ILs can be repeatedly recycled for CO2 uptake (four cycles) with no observed loss of absorption rate and absorption capacity.
 | ||
| Scheme 5 Structures of amino acids-based tetraalkyl ammonium ionic liquids.51,52 | ||
And then, Zhang and coworkers prepared fifteen novel AAILs by the combination of several asymmetric tetraalkylammonium cations with four amino acid anions ([Gly], [L-Ala], [β-Ala] and [Val]) (Scheme 5).52 The asymmetry of the tetraalkylammonium cations is shown to have a significant influence on the viscosity of the ILs being composed of AA anions, especially for the four triethylbutylammonium ([N2224])-based ILs that have viscosities of lower than 60 mPa s, with the lowest being only 29 mPa s. The mole uptake of CO2 per mole [N2224][L-Ala] is found to approach 0.5, equivalent to 0.326 mole fraction of CO2 in the IL, in 20 min at 40 °C.
Absorption/desorption of CO2 by ILs where both cation and anion are from renewable materials has also been developed adopting (2-hydroxyethyl)-trimethyl-ammonium (S)-2-pyrrolidine-carboxylic acid salt [Choline][Pro] (50 °C) (Scheme 6), and a [Choline] [Pro]/polyethylene glycol (PEG200) mixture (35 °C) as absorbents.53 For the neat IL, the time required for absorption is longer than 240 min because the IL became very viscous after absorbing enough CO2, while only 50 min is needed for the mixture with a [Choline][Pro] to PEG200 weight ratio of 1:1.
![Schematic illustration of the synthesis of the IL [Choline][Pro].53](/image/article/2011/RA/c1ra00307k/c1ra00307k-s6.gif) | ||
| Scheme 6 Schematic illustration of the synthesis of the IL [Choline][Pro].53 | ||
While amine-functionalized ILs are used for CO2 uptake, 1:2 stoichiometry arising from amine deprotonation of a carbamic acid intermediate is atom inefficient. Gurkan et al. synthesized two amino acid-based ILs, including trihexyl (tetradecyl) phosphonium prolinate ([P66614][Pro]) and methioninate ([P66614] [Met]), that can react with CO2 in a ratio of one CO2 per amine (1:1 stoichiometry) (Scheme 7) by termination of the reaction sequence at the formation of carbamic acid.11 In the FTIR spectrum of [P66614][Pro] before and after reaction with CO2 (at 0.2 bar) (Fig. 2), the prolinate N–H stretch at 3290 cm−1 disappears when CO2 reacts with the IL. There is no evidence for ammonium formation, because if ammonium ions are present, absorption peaks for the ammonium group between 3000 and 2800 cm−1 and combination bands in the 2800–2000 cm−1 region would be observed. A new band centered at 1689 cm−1 corresponds to the new COOH moiety.
![IR spectrum of [P66614][Pro] before and after reaction with CO2. The inset shows the disappearance of the N–H stretch. Reprinted with permission from ref. 11. Copyright 2010 American Chemical Society.](/image/article/2011/RA/c1ra00307k/c1ra00307k-f2.gif) | ||
| Fig. 2 IR spectrum of [P66614][Pro] before and after reaction with CO2. The inset shows the disappearance of the N–H stretch. Reprinted with permission from ref. 11. Copyright 2010 American Chemical Society. | ||
![Reaction schematics of CO2 with [P66614][Met] (top) and [P66614][Pro] (bottom).11](/image/article/2011/RA/c1ra00307k/c1ra00307k-s7.gif) | ||
| Scheme 7 Reaction schematics of CO2 with [P66614][Met] (top) and [P66614][Pro] (bottom).11 | ||
Combinatorial chemistry and click chemistry have been applied to synthesize sixty-three kinds of TSILs from primary amines, sultones, and quaternary ammonium hydroxides, bearing secondary amine groups in the anions for CO2 capture (Scheme 8), among which sixteen are intrinsically resin-, plastic-, or gel-like and could capture CO2 in the those forms.54
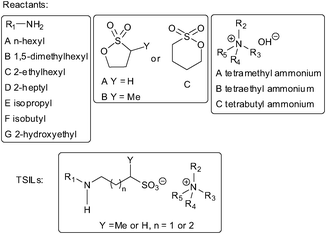 | ||
| Scheme 8 Structures of reactants and the TSILs for CO2 capture.54 | ||
Superbase-derived ionic liquids for CO2 capture
Recently, an innovative CO2 capture system based on the formation of amidinium or guanidinium alkylcarbonate salts with good reactivity and high absorption capacity has shown interesting promise.55–59 This CO2 capture system consists of an alcohol and an amidine (or guanidine) superbase. Compared with aqueous amine solution systems, the low specific heat and reduced hydrogen bonding in alkylcarbonate salts could result in a less energy intensive CO2 release.60 However, one key drawback associated with this method for the capture of CO2 could be the volatile nature of its molecular constituents (i.e., alcohols and/or base), probably leading to the loss of organic solvents. Based on the previous reports, Dai et al. have reported CO2 sorption behavior and selective absorption of CO2/N2 performance using four different integrated systems consisting of 1:1 mixtures of proton donor and an appropriate superbase.12,61–63
The first strategy they adopted is to incorporate hydroxyl group into non-volatile room temperature ionic liquids (RTILs) cations to serve as the proton donor, the issues associated with the volatilization of the alcohol can be mitigated accordingly (Scheme 9).61 Superbases with high proton affinities would play a key role as proton acceptors, thereby providing a thermodynamic driving force for CO2 capture.
 | ||
| Scheme 9 Chemical structures and their designations of the superbases and hydroxyl-functionalized ionic liquids.61 | ||
A CO2 to 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) molar ratio of 1.04 can be achieved when the TSIL [Im21OH][Tf2N] is used, which is at least 20 times greater than that in neat [Im21OH][Tf2N].64 In addition, absorption is nearly complete within the first 10 min of CO2 exposure for this system. The release of CO2 can essentially finish within 15 min at 120 °C and CO2 absorption into and release from the [Im21OH][Tf2N]-DBU solution can be repeatedly recycled for three times with only a slight loss of absorption capability. CO2 could undergo a reaction with [Im21OH][Tf2N] and DBU to form the liquid amidinium alkylcarbonate salt during the absorption reaction of CO2 (Scheme 10).
 | ||
| Scheme 10 Reaction mechanism of CO2 absorbed by RTIL-superbase system.64 | ||
Equimolar CO2 absorption capacity can be reached when either a bicyclic amidine (DBU) or a guanidine (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD)) superbase is used, whereas CO2 absorption in the presence of a phosphazene-type superbase is reduced by roughly 20% and 50% in the case of BEMP and EtP2 (dma), respectively, which may be related in part to the excessive viscosity of the latter RTIL-superbase systems and their corresponding alkylcarbonate salts.
It is well known that the C-2 proton of imidazolium-based ILs has a weak acidity. Its pKa in DMSO is about 24.0.65 It can also be utilized to replace volatile alcohol or amine to serve as weak proton donor for CO2 capture.62 Six different imidazolium ILs and four superbases are selected to study CO2 capture performance (Scheme 11).
 | ||
| Scheme 11 Chemical structures and their designations of the superbases and imidazolium ionic liquids.62 | ||
The influence of different ILs on CO2 absorption is significant. Among them, the absorption of CO2 by both [BMIm][Tf2N]-DBU and [EMIm][Tf2N]-DBU approaches 1.0 mol per mole of IL in 30 min. However, the absorption molar ratios of CO2 to IL are reduced dramatically to 0.80 or 0.75, respectively, when [BMIm][BF4] and [BMIm][PF6] are used probably due to the increased viscosities of these systems. In contrast, when pyridinium-based IL [BPy][Tf2N] without the acidic proton is used, only physical interaction between this system and CO2 is observed (0.29). Equimolar CO2 could be captured by employing different superbases except TMG. The absorption capacity of CO2 in the [BMIm][Tf2N]-TMG system is reduced to 0.49 mol per mole of superbase because of the formation of stronger hydrogen bonding between carboxylate salt and [TMGH+].60
During the absorption of CO2 by [BMIm][Tf2N]-MTBD system, CO2 is assumed to react with imidazolium-based ILs and superbases to form the liquid amidinium carboxylate salt (Scheme 12). After CO2 bubbling, the new band for [bmim][Tf2N]-MTBD + CO2 at 1669 cm−1 can be assigned to carboxylate stretches.66 Furthermore, the 13C NMR spectrum shows the carboxylate peak at 155.6 ppm. The release of CO2 proceeds rapidly for the [BMIm][Tf2N]-DBU system at 80 °C under bubbling N2, and the CO2 release is essentially complete within 20 min. In addition, the process of CO2 capture by imidazolium-based IL-superbase systems is reversible.
![Reaction mechanism of CO2 absorption by [BMIm][Tf2N]-MTBD.62](/image/article/2011/RA/c1ra00307k/c1ra00307k-s12.gif) | ||
| Scheme 12 Reaction mechanism of CO2 absorption by [BMIm][Tf2N]-MTBD.62 | ||
Dai et al. have also reported an efficient carbon capture system based on a diverse class of anion-functionalized protic ionic liquids (PILs).12 The essence of this protocol is to use a very strong base (superbase or proton sponge) to directly deprotonate weak proton donors, such as fluorinated alcohols, imidazoles, pyrrolidinones or phenols (Scheme 13), obtaining PILs with low melting points, which are capable of reversibly capturing CO2 with high capacity (more than 1 mol per mol IL). A molar ratio of CO2 to PILs of 1.13 could be achieved when [MTBDH+][TFE−] is used; [MTBDH+][HFPD2−] shows a higher CO2 capacity of more than 2.0 mole per mole PIL (2.04) because of the presence of two CO2-reactive groups. The absorption is almost completed within 5 min for [MTBDH+][TFE−], whereas in the case of [MTBDH+][Im−], ca. 30 min is needed to reach sorption equilibrium. The rapid absorption is related to their low viscosities. For example, the viscosity of [MTBDH+][TFE−] and [MTBDH+][Im−] are 8.63 cP, 31.85 cP, respectively. In contrast, the chemically unreactive PIL, such as [MTBDH+][Tf2N−], has a capacity of only 0.02 mol per mole PILs. CO2 reacts with PILs to form a liquid carbonate, carbamate or phenolate salt in terms of mechanistic consideration (Scheme 14). After CO2 bubbling, a new band for [MTBDH+][ImC−] at 1696.4 cm−1 can be assigned to carbamate stretches. The release of CO2 by bubbling N2 at 80 °C for [MTBDH+][ImC−] is almost completed in 10 min. The processes of CO2 absorption by PILs are completely reversible, and the PILs show a slight loss in capacity after multiple absorption-desorption cycles.
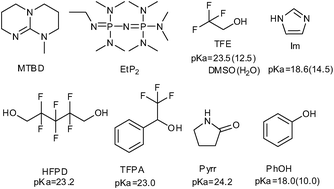 | ||
| Scheme 13 Selected superbases, fluorinated alcohols, imidazole (Im), pyrrolidone (Pyrr), and phenol used as building blocks of superbase-derived PILs. pKa values in DMSO (values given in brackets are in H2O).12 | ||
 | ||
| Scheme 14 CO2 absorption by the anions of superbase-derived protic ionic liquids.12 | ||
Recently, they have further reported a strategy to tune the enthalpy of CO2 absorption by tunable basic ILs prepared by neutralizing eight kinds of weak proton donors with different pKa values in DMSO ranging from 19.8 to 8.2 with phosphonium hydroxide (Scheme 15).63 The absorption capacity of CO2 can be enhanced dramatically from 0.17 to 0.95 mol CO2 per mole IL when the pKa of the anion in DMCO increases from 11.9 ([P66614][Bentriz]) to 13.9 ([P66614][Triz]). But the increase of CO2 absorption capacity is not remarkable (from 0.95 to 1.02) when the pKa of the anion in DMSO further increases from 13.9 to 19.8 ([P66614][Pyr]). The absorption of CO2 is almost completed in 10 min with all of these basic ILs, which is substantially faster than with conventional amino-functionalized ILs. The results in Fig. 3 show a rough linear relationship between absorption enthalpy and the pKa value. It reveals that the enthalpy of CO2 absorption by these basic ILs can be quantitatively tuned by varying the basicity of the IL. CO2 absorption into and release from [P66614][Triz] can be repeatedly cycled for 25 times with high absorption capacity and rapid absorption rate.
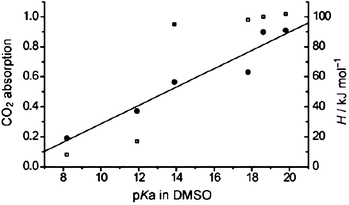 | ||
| Fig. 3 The relationship between CO2 absorption capacity (□), absorption enthalpy (ΔH, ●), and the pKa value of the anion in DMSO. The linear fit for the relationship between △H and pKa of the anion in DMSO is shown; R2 = 0.930. Reprinted with permission from ref. 63. Copyright 2011 John Wiley and Sons. | ||
 | ||
| Scheme 15 Structure of anion and cation in tunable basic ILs for CO2 capture. Values given in brackets are in H2O.63 | ||
Several single molecules containing both a CO2-capturing group and a basic functional group (amidine-alcohol, guanidine-alcohol, or diamine) have been synthesized to serve as switchable CO2 absorbents.67 The reaction of anhydrous 1a with CO2 leads to the formation of a highly viscous hazy liquid (1b) (Scheme 16). The neat 13C NMR spectrum of 1b shows the guanidinium carbon at 161.3 ppm as well as the alkylcarbonate peak at 152.2 ppm. The IR spectrum of 1b gives new strong bands at 1590 cm−1, 1403 cm−1 and 1275 cm−1, being assigned to the (CO) of the carbonate. Medium intensity N–H stretching bands at 3160 cm−1 are also observed. Gravimetrically, 0.502 g (3 mmol) of 1a absorbed 0.130 g (3 mmol) of CO2 corresponds to 20.6% CO2 by weight. However, the zwitterion 1b does not give off gas, rather it slowly cyclizes into a cyclic carbonate.
 | ||
| Scheme 16 Reactions of alkanol-guanidines with CO2.67 | ||
Amidines are slightly less basic than guanidines but still sufficiently basic to serve as the basic group in a zwitterionic liquid precursor (Scheme 17). Upon the reaction of 4a with CO2, the mass of the liquid phase increases by 20%, and the theoretical mass increase should be 25%.

Liquid secondary amines represent another kind of switchable solvent, wherein CO2 is bound directly to the nitrogen atom in a carbamate salt. The liquid diamines of the structure RHN(CH2)nNHR where n = 2 (R = Me), n = 3 (R = Me, Et, i-Pr), and n = 6 (R = Me, Et, Bu) all give solids upon reaction with CO2 (Scheme 18).
 | ||
| Scheme 18 Reactions of secondary amines with CO2.67 | ||
Bubbling CO2 through neat liquid N,N′-trimethyl-1,3-propanediamine (6a) (diamine containing one secondary and one tertiary amine) could get the zwitterionic product [Me2HN+CH2CH2CH2N(Me)CO2−] (6b) (Scheme 19). The IR spectrum of the solid shows the disappearance of the N–H band at 3298 cm−1 and the appearance of a band at 1545 cm−1, which could be characteristic of a carbamate anion.
 | ||
| Scheme 19 Reactions of diamine containing one secondary and one tertiary amine with CO2.67 | ||
Chemical absorption of CO2 using ionic liquids as solvents
Solutions of RTILs and commercially available amines are found to be effective for the capture of CO2 as carbamate salts.68 Primary alkanolamine (monoethanolamine, MEA) is dissolved in 7a and 7b (Scheme 20) as 50:50 (mol/mol) solutions (16% v/v MEA), containing one primary amine per ion pair, analogous to amine-substituted TSILs.10 Solutions are exposed to CO2 at ∼1 atm and 40 °C, and the reaction is complete after 25 min to give an insoluble MEA-carbamate precipitate that helps to drive the capture reaction (CO2 capture capacity is 1 mole of CO2 per 2 moles MEA). Because RTILs with Tf2N anions have relatively low viscosities, the absorption time can be only ~10% of that for a highly viscous TSIL. CO2 can be rapidly decomplexed from MEA-carbamate in 7a by heating the resulting mixture to 100 °C and/or through reducing the pressure. At 40 °C and a final CO2 pressure of 4 kPa, an equimolar 7b-diethanolamine (DEA) solution contains 0.29 mole of CO2 per mole of DEA. CO2 and DEA are in equilibrium with DEA-carbamate at low pressure and the capture does not go to completion.
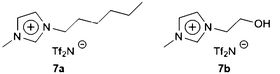 | ||
| Scheme 20 General structures of (7a) imidazolium-based RTILs and (7b) amine-substituted “task-specific” ionic liquids.68 | ||
Chitin and chitosan (CS) as substitutes for amino-functionalized synthetic polymers dissolving in the IL 1-butyl-3-methyl-imidazolium chloride ([BMIm]Cl), which have a strong ability to disrupt hydrogen bonds, can be utilized for capturing and releasing CO2 (Scheme 21).69 Chitosan, a natural biomaterial, is an N-deacetylated product of chitin, which is the second abundant natural polymer after cellulose and has a similar structure to cellulose.70 [BMIm]Cl and chitosan powder has almost no ability to absorb CO2. At equilibrium, the 10 wt% chitosan/IL and chitin/IL solutions exhibit approximately 8.1% and 3.8% CO2 fixing efficiency, respectively. Due to the fact that there exist many amino groups in chitosan and none in chitin, physically dissolved CO2 could be responsible for the CO2 sorption capacity of chitin/IL solution; whereas, both physically dissolved CO2 and chemically bound CO2 are responsible for the effective CO2 sorption capacity of chitosan/IL solution. It takes 180 min for chitosan/IL solution to reach 98% sorption capacity, and for chitin/IL solution to get 95% sorption capacity. Taking the 10 wt% chitosan/IL solution as an example, the CO2 can be reduced to a 1.4% level in 30 min at 100 °C, and is completely released under vacuum.
 | ||
| Scheme 21 Reversible covalent chemistry between CO2 and amines linked to the chitosan polymer chain.69 | ||
Task-specific ionic liquid-catalyzed conversion of carbon dioxide into value-added chemicals/fuels
Cyclic carbonate synthesis
One of the few commercial routes using CO2 as a raw material can be the insertion of CO2 into epoxides to afford the 5-membered cyclic carbonates (Scheme 22), such as ethylene carbonate (EC) and propylene carbonate (PC), which can be served as polar aprotic solvents, electrolytes in secondary batteries, valuable precursors for polycarbonate and polyurethane materials and intermediates in organic synthesis.71 In the past decades, numerous heterogeneous catalysts have been proposed for this reaction, such as metal oxides, oxychlorides, Cs-loaded zeolites and alumina, active species supported by natural or synthesized polymers, silica, zeolites and other materials. In addition, the cycloaddition reaction also proceeds smoothly using homogeneous catalysts including amines and phosphines, alkali metal halides and onium salts, organometalic compounds, CO2 adducts of N-heterocyclic carbenes and especially ILs.19,21 Among these, alkali metal halides and ammonium salts have already been used as efficient homogeneous catalysts for industrial production. In particular, ILs are considered to be one of the most efficient catalysts for the cycloaddition reaction. | ||
| Scheme 22 Cycloaddition reaction of CO2 to epoxide. | ||
Homogeneous ionic liquid catalysts
A straightforward method for chemical fixation of CO2 onto epoxides has been achieved by simply dissolving the substrate in molten tetrabutylammonium bromide (TBAB) (120 °C) or tetrabutylammonium iodide (TBAI) (60 °C) in the presence of CO2 at atmospheric pressure.72 Once the reaction is completed, pure cyclic carbonates can be isolated by vacuum distillation or extraction with ethyl acetate, in which the IL (TBAB/TBAI) is insoluble. This procedure allows the recycling of the ammonium salt. A plausible mechanism for this reaction (Scheme 23) would be the ring opening of the epoxide by means of a nucleophilic attack by the bromide ion, which can lead to an oxy anion species affording the corresponding cyclic carbonate after reaction with CO2. | ||
| Scheme 23 Plausible mechanism for the cycloaddition of epoxides and CO2.72 | ||
The first successful quantitative imidazolium ILs-catalyzed synthesis of PC was reported by Peng and Deng, using 2.5 mol% of 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm][BF4]) 8a under electro-catalytic conditions (Scheme 24), although the reaction is carried out at 2.5 MPa CO2 pressure and 110 °C for 6 h with a TOF of just 6.6 h−1.73
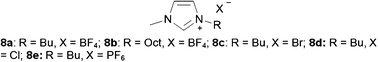 | ||
| Scheme 24 Structure of imidazolium ionic liquids. | ||
The mixture of ILs and supercritical CO2 (scCO2) may offer new opportunities for the development of the reaction due to the complete miscibility of CO2 in the ILs under high pressure conditions. Ikushima et al. reported the application of a scCO2/1-octyl-3-methylimidazolium tetrafluoroborate ([OMIm][BF4]) 8b (Scheme 24) biphasic system to CO2 fixation. The yield and selectivity of PC of nearly 100% can be achieved within 5 min with a high turnover frequency (TOF) value (= 517 h−1), leading to a 77-fold faster rate of reaction than previously reported.73 BF4− is found to be the most effective anion and yield of PC is increased with lengthening alkyl chain (from C2 to C8) due to an increase in the solubility of CO2 and propylene oxide (PO) in IL phase.74
The role of Lewis acid species such as ZnBr2 with ILs as catalysts is investigated and large improvements in the yields can be realized, although high-pressure conditions are still necessary.75 The single crystal X-ray diffraction analysis of an active species (1,3-dimethylimidazolium)2ZnBr2Cl2 shows that two 1,3-dimethylimidazolium cations are paired with a dibromodichloro zincate dianion.76 A similar system combining zinc salts with [BMIm][X] (X = Cl, Br, BF4, PF6) 8a,8c–8e (Scheme 24) has been utilized for the addition of CO2 to PO.27 The best result in terms of product yield and TOF is obtained by using ZnBr2 with [BMIm][Br] 8c which give 98% yield and a TOF of 5580 h−1. Reactions are carried out at 100 °C for 1 h using a CO2 pressure of 1.5 MPa. Other epoxides, including epichlorohydrin, 1,2-epoxybutane and styrene oxide are also tested, giving good results with the exception of cyclohexene oxide which gives a yield of only 34% (TOF: 1206 h−1). A catalytic cycle is also proposed (Scheme 25) which involves activation of the epoxide by zinc and subsequent ring-opening with Br−. CO2 could then insert into the Zn-O bond with ring-closure giving the cyclic carbonate. Using BF4 or PF6 as the IL counterion (8a,8e) results in minimal catalytic activity, thus proving that the nucleophilic nature of the anion could be crucial for the reaction to occur.
![Catalytic cycle for ZnX2 facilitated addition of CO2 to epoxides in the presence of the ionic liquid [BMIm]Br.27](/image/article/2011/RA/c1ra00307k/c1ra00307k-s25.gif) | ||
| Scheme 25 Catalytic cycle for ZnX2 facilitated addition of CO2 to epoxides in the presence of the ionic liquid [BMIm]Br.27 | ||
The conversion of styrene oxide (SO) to styrene carbonate (SC) could be more difficult due to the lower reactivity of the β-carbon atom as compared with PO and ethylene oxide (EO). Catalyst system comprising ZnBr2 and an IL [BMIm]Cl 8d (Scheme 24) can afford 93% yield with 100% selectivity for SC at a low reaction temperature of 80 °C for 1 h.77,78 Several metal halides as a co-catalyst with 8d are screened for the coupling reaction of SO and CO2, and the order of activity are found to be Zn2+ > Fe3+ > Fe2+ > Mg2+ > Li+ > Na+.
Hexabutylguanidinium ILs are more effective than the imidazolium salt analogues.79 A catalyst system consisting of hexabutylguanidinium salt ILs and ZnBr2 for chemical fixation of CO2 onto epoxides is employed.80 The optimum reaction temperature and CO2 pressure in this catalytic process would be 130 °C and 3.0 MPa. TOF values as high as 6.6 × 103 h−1 for SO and 1.01 × 104 h−1 for epichlorohydrin are obtained.
A series of tetrahaloindate(III)-based ILs with a general formula of [Q][InX3Y](Q = imidazolium, phosphonium, ammonium and pyridium; X = Cl, Br, I; Y = Cl, Br) are used to catalyze the reaction of CO2 with a variety of epoxides (Scheme 26).81 Tetrahaloindate(III)-based ILs are found to exhibit high catalytic activities and evidence is presented that supported the significant role of H-bonding interactions in the [Q][InX3Y]-catalyzed coupling reactions. The plausible reaction mechanism based on the 1H and 13C NMR studies for the formation of PC is presented in Scheme 27.
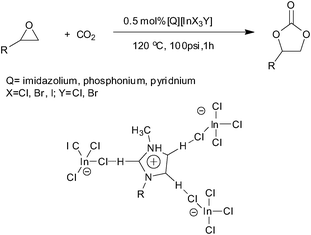
 | ||
| Scheme 27 Reaction mechanism for the formation of propylene carbonate catalyzed by tetrahaloindate(III)-based ionic liquids.81 | ||
A series of hydroxyl-functionalized ILs (HFILs) (Scheme 28) has been synthesized and characterized.28 Efficient reactivity and reusability toward the coupling of epoxide and CO2 are attained. Highest activity and selectivity are achieved in the presence of 1-(2-hydroxyl-ethyl)-3-methylimidazolium bromide (HEMImB) in comparison with other similar catalysts investigated. Under the optimized reaction conditions (catalyst loading, 1.6 mol%; CO2 pressure, 2.0 MPa; 125 °C, 1 h), yield and selectivity of PC are 99% and 99.8% respectively. The mechanism shows that the OH group and the Lewis basic site bromide anion of HFIL simultaneously attack the different parts of epoxide (Scheme 29). The coordination of the H atom with the O atom of epoxide through a hydrogen bond could result in the polarization of C–O bonds, so the ring of the epoxide can be opened easily.
 | ||
| Scheme 28 Structures of hydroxyl-functionalized ionic liquids (HFILs).28 | ||
 | ||
| Scheme 29 Proposed mechanism for the HFILs-catalyzed cycloaddition reaction of CO2 with epoxides.28 | ||
Carboxylic acid groups can also accelerate the ring-opening reaction of epoxides through forming hydrogen bonds. A series of betaine-based salts containing quaternary ammonium ion and carboxylic acid group are synthesized by direct protonation of anhydrous betaine using different Brønsted acids (Scheme 30).82 The order of the catalytic activity of these salts for the synthesis of cyclic carbonates via cycloaddition reaction of CO2 with epoxides is found to be HBetI > HBetCl > HBetBr. Under the conditions of 8 MPa (CO2 pressure), 140 °C, 8 h with the catalyst (HBetI) loading of 2.5 mol%, yield of PC is 98%.

Our group has developed a series of easily prepared Lewis basic ILs (Scheme 31) for cyclic carbonate synthesis from epoxide and CO2 at low pressure without utilization of any organic solvents or additives.21 Interest in these salts stems from their facile preparation from commercially available and relatively inexpensive starting materials, gratifyingly thermal behaviour, air/water stability. More importantly, the presence of the tertiary nitrogen in the cation has the potential to form the carbamates species with CO2, which can be considered as an activated form of CO2. On the other hand, epoxide activation could be attained by a hydrogen bond formed in situ between the oxygen of the epoxide and the proton attached to the ammonium nitrogen atom of the IL. The results reveal that catalytic efficiency decreases in the order of HDBU+ > HTBD+ ∼ OMIm+ > C4DABCO+∼C8DABCO+ > BMIm+ > HHMTA+. Indeed, the IL [HDBU]Cl displays the highest catalytic activity for conversion of CO2 and almost quantitative yield (97%) together with excellent selectivity (>99%) of PC is obtained under optimized reaction conditions ([HDBU]Cl, 1 mol%; CO2 pressure, 1 MPa; 140 °C; 2 h).
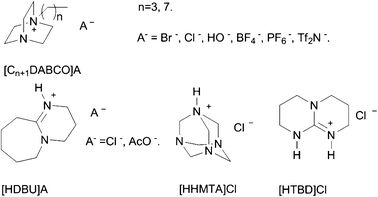 | ||
| Scheme 31 Lewis basic ionic liquids used in this study as catalysts for the cycloaddition reaction of CO2 with epoxides.21 | ||
Electrochemical activation of CO2 is performed in ILs (Scheme 32),83 and electrocatalytic cycloaddition of CO2 to epoxides in RTILs as reaction media without any additional supporting electrolyte and catalyst could be conducted with high to excellent performances under mild conditions.
 | ||
| Scheme 32 Electrocatalytic cycloaddition of CO2 to epoxides.83 | ||
ILs can also be used as solvents to produce unsaturated cyclic carbonates. Reactions of propargylic alcohols with CO2 are performed in a [BMIm][PhSO3]/CuCl catalytic system to produce the corresponding α-methylene cyclic carbonates.22 High yield (>95%) can be achieved over catalyst CuCl/CuBr/CuI using [BMIm][PhSO3] as solvent at 1 MPa CO2 pressure, 120 °C, 8 h. In comparison with the results obtained from organic solvents, the catalytic efficiency and yields could be greatly enhanced in ILs. For example, 97% of yield is obtained when the reaction of 2-methyl-3-butyn-2-ol with CO2 proceeds in IL [BMIm][PhSO3] for 8 h, whereas only 60% yield is given in DMF for 24 h.
Heterogeneous ionic liquid catalysts
Although ILs have been reported to be one of the most efficient catalysts for CO2 fixation to produce cyclic carbonate from epoxides, application of homogeneous IL catalysts in industry could be limited due to the complicated separation procedure from the reaction mixture. On the other hand, proper use of heterogeneous catalysis in CO2 conversion may afford enhancement of the reaction rate, control of selectivity, increasing catalyst lifetime and facilitating separation. For example, Si-OH groups on the silica surface can act as a weak acid to activate the epoxide, and silica as a support has synergistic effect in promoting the reaction.84Our group has reported a silica-supported quaternary ammonium salt for the first time as a recyclable and efficient catalyst for the synthesis of PC from PO and CO2 under supercritical conditions, which requires no additional organic solvents either for the reaction or for the separation of product.85 Silica-supported catalysts possess slightly higher catalytic activity for PC synthesis than the unsupported counterparts. n-Bu4NBr/SiO2 exhibits high catalytic activity and excellent selectivity for PC synthesis at 8 MPa, 150 °C, 8 h. We have also synthesized amorphous silica supported [BMIm]X (X = BF4, PF6, Br) (Scheme 24) (denoted by [BMIm]X/SiO2) for the synthesis of cyclic carbonates from epoxides and CO2 under supercritical conditions.86 The obtained heterogeneous catalysts are found to exhibit high activity for the solvent-less synthesis of cyclic carbonates, and high yields (>93%) with excellent selectivity (>97%) are obtained at temperature of 160 °C under 8 MPa. In addition, the catalyst can be easily recovered and reused four times with slight loss of its catalytic activity.
Silica-supported hexaalkylguanidinium chloride (Scheme 33) is utilized as an effective catalyst for CO2 fixation to carbonate without any solvent under mild reaction conditions (4.5 MPa, 120 °C, 4 h),79 and it could be recycled easily at least five further times without any obvious decrease in its catalytic activity after simple filtration.

The catalytic activity of phosphonium salts towards cyclic carbonate synthesis from PO and CO2 has been enormously enhanced by their immobilization onto silica, but itself almost has no catalytic activity.87 It is noteworthy that the pseudo-first order rate constant of SiO2-C3H6-P(n-Bu)3I normalized to a phosphorus atom is about 300 times larger than that of P(n-Bu)4I. An almost quantitative amount of PC (PC yield: 99.7%) can be produced using SiO2-C3H6-P(n-Bu)3I as catalyst at 100 °C and 10 MPa of CO2 within 1 h.
Different mesoporous silica-supported imidazolium ILs 9a–f are prepared as effective heterogeneous catalyst for the solventless synthesis of cyclic carbonate from allyl glycidyl ether and CO2 (Scheme 34).88–92 The supported ILs are characterized using XRD, BET, FT-IR, EA (elemental analysis), SEM, CP 13C-, 29Si-MAS-NMR and TG/DTG. The effects of temperature, catalyst amount, influence of alkyl chain length, and variation of CO2 pressure are examined.
 | ||
| Scheme 34 Cycloaddition of allyl glycidyl ether and CO2 catalyzed by silica supported ionic liquids.88–92 | ||
It has been reported that the stability of counter anions (ring-opened epoxide) increases in the presence of large, delocalized counter cations with resonance forms.93 Silica-supported 4-aminopyridinium halides are chosen as acid–base bifunctional catalysts for the transformation of epoxides to cyclic carbonates under atmospheric pressure of CO2.84 Among the catalysts shown in Scheme 35, SiO2-1(I) is the most efficient catalyst, affording 91% conversion of SO and SC in 89% yield under conditions of CO2 (1 atm), 100 °C and 20.5 h. The catalytic activity of anions decreases in the order of I− > Br− > Cl−. The recovered catalyst SiO2-1(I) could be reused at least three times without any appreciable loss of activity and selectivity. SiO2-supported catalysts show higher performances than the physical mixtures containing SiO2. What is more, the reaction of both cis- and trans-2,3-epoxybutane using SiO2-1(I) catalyst gives retentions of configuration: cis:trans ratio of carbonate product are 91:9 for cis-2,3-epoxybutane and 3:97 for trans-2,3-epoxybutane, respectively.
Insoluble ion exchange resins, one type of polystyryl supported catalysts containing an ammonium salt or amino group, and the polar macroporous adsorption resin, are efficient and reusable heterogeneous basic catalysts for the synthesis of PC from PO and CO2 under scCO2 conditions (100 °C, 8 MPa), which requires no additional organic solvents either for the reaction or for the separation of product.94 A quantitative yield (97.4%) together with excellent selectivity (99.4%) of PC is obtained catalyzed by ion exchange resin (D201). The catalyst can be easily recovered and reused without significant loss of its catalytic activity. The process represents a simple, ecologically safer, cost-effective route to cyclic carbonates with high product quality, as well as easy product recovery and catalyst recycling.
Han and co-workers supported the IL active species on a highly cross-linked polymer matrix to prepare an active and insoluble catalyst.95 3-Butyl-1-vinylimidazolium chloride ([VBIm]Cl) is copolymerized with the cross-linker divinylbenzene (DVB) to prepare a highly cross-linked polymer-supported IL (PSIL), in which [VBIm]Cl is covalently anchored on DVB-cross-linked polymer matrix (Scheme 36). The catalytic activity of the PSIL for the reaction of CO2 and PO is studied. Under optimal reaction conditions (6 MPa CO2, 110 °C, 7 h), PC yield is 97.4%. There is no considerable decrease in the yield of PC for the five repeated runs, indicating that the catalyst is not only insoluble in the reaction mixture but is also very stable.
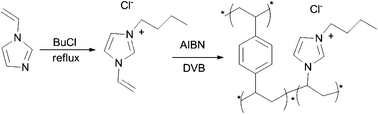 | ||
| Scheme 36 Synthesis of the cross-linked-polymer-supported ionic liquid. AIBN: azobis(isobutyronitrile).95 | ||
The catalytic activity of polyaniline-HX (X = I, Br, Cl) (PANI-HI, PANI-HBr, PANI-HCl) has also been investigated for the cycloaddition of CO2 to PO to produce PC.96 Polyaniline (PANI) is one of the most useful conducting polymers due to its facile synthesis, environmental stability and simple acid/base doping/dedoping chemistry (Scheme 37). It is shown that all the PANI salts are active for the reaction, and PANI-HI is most active and selective. Complete conversion can be achieved in 3 h for epichlorohydrin and 6 h for PO at 115 °C and 5 MPa. With PO as the substrate, the reusability of PANI-HI is evaluated and no loss of catalytic activity is detectable after the catalyst has been reused five times.
 | ||
| Scheme 37 Structure of polyaniline and its acid/base doping/undoping process.96 | ||
CS is the N-deacetylated derivative of chitin which is one of the most abundant biopolymers next to cellulose and a by-product of fishing industry. Besides the bio-compatibility, biodegradability and non-toxicity, CS can be easily chemically or physically modified which made it an excellent supporting material of catalyst. We designed and synthesized a functionalized biopolymer-chitosan-supported quaternary ammonium salt (CS–N+R3X−) (Scheme 38), which is shown to be a single-component active catalyst for the cycloaddition reaction of epoxides and CO2.97 The catalyst can be easily recovered by filtration and reused for at least five times without obvious loss of its catalytic activity.
 | ||
| Scheme 38 Preparation of the quaternary ammonium salts covalently bound to chitosan.97 | ||
Poly(ethylene glycol)s (PEGs) are a family of water-soluble linear polymers formed by the interaction of ethylene oxide with water, ethylene glycol, or ethylene glycol oligomers. Our interest in PEGs stems from its distinctive properties, such as inexpensive, thermally stable, almost negligible vapor pressure, toxicologically innocuous and environmentally benign characterization. More importantly, PEG could be regarded as a CO2-philic material. In other words, “CO2-expansion” effect could lead to changes in the physical properties of the liquid phase mixture including lowered viscosity and increased gas/liquid diffusion rates.98 We used one PEG (M.W. = 6000) derivative, which is covalently bound to a quaternary ammonium salt, as an efficient catalyst for CO2 fixation with PO without any organic additive under supercritical conditions to afford PC in quantitative yield and excellent selectivity.99 Furthermore, the PEG6000-supported catalyst can be readily recovered by simple filtration and reused over five times without appreciable loss of activity. The reaction of PO with CO2 in the presence of 0.5 mol% PEG6000(NBu3Br)2 affords PC in 98% yield together with 99% selectivity at 120 °C, 8 MPa, 6 h.
We envisions that a functionalized-PEG, hexaalkyl guanidinium bromide being covalently tethered to PEG6000 (M.W. = 6000), could be utilized as an active catalyst for the synthesis of cyclic carbonates from CO2 and epoxide (Scheme 39).100 The catalyst is found to be applicable to a variety of terminal epoxides, providing the corresponding cyclic carbonates in high yields and selectivity (Scheme 40). It has been found that there is a pronouncedly cooperative effect between the catalyst part and the support part. Moreover, the catalyst is able to be reused for four times with retention of high catalytic activity and selectivity.
 | ||
| Scheme 39 The synthesis of PEG6000-supported guanidinium bromide.100 | ||
 | ||
| Scheme 40 Synthesis of cyclic carbonates catalyzed by PEG6000-supported hexaalkylguanidinium bromide.100 | ||
In recent years, metal–organic frameworks (MOFs), which are formed by copolymerization of organic molecules with metal ions or metal ion clusters, have received much attention because of their zeolite-like properties, such as high internal surface area and microporosity, well-ordered porous structures and high absorption capacity. Han's group conducted the coupling reaction of CO2 and epoxides using MOF-5 [Zn4O(BDC)3 (BDC = benzene-1,4-dicarboxylate)], which is copolymerized by a Zn4O cluster with BDC through octahedral arrays, as the heterogeneous catalyst in the presence of quaternary ammonium salts.101 MOF-5/n-Bu4NBr system could convert various epoxides to the corresponding cyclic carbonates effectively under mild conditions (n-Bu4NBr, 2.5 mol%; MOF-5, 0.1 g; CO2 pressure, 0.1 MPa; reaction temperature, 50 °C) with a SC yield of 92% within 15 h.
A general approach for the covalent immobilization of glycidyl-group-containing ILs on organic and inorganic supports with functional surfaces (Scheme 41) has been proposed, based on the fact that the glycidyl group can actively react with almost all nucleophilic, electrophilic, neutral, and free-radical species.102 By using polymer spheres with amino- and carboxyl- group-functionalized surfaces as organic supports and silica (including SBA15 and silica gel) with amino groups attached as inorganic supports, the IL 1-glycidyl-butylimidazolium chloride is successfully grafted onto these polymer and silica supports, respectively, through reactions between the glycidyl group in the IL and the polar groups on the support surfaces. The activities of these resultant polymer- and silica- based catalysts are investigated for CO2 cycloaddition reactions with epoxides. In particular, the polymer supports generated synergistic effects with the IL in the coupling reaction of CO2 with PO, and the TOF could reach about 1700 h−1 when the IL attached to the NH2-functionalized polymer is used as the catalyst.
 | ||
| Scheme 41 General approach for the covalent immobilization of a glycidyl-group-containing IL on supports functionalized with active groups through oxirane-ring-opening reactions.102 | ||
Polymeric nanoparticles (NPs) have received increasing attention due to their potential applications as nanocarriers for catalysts, molecules with electronic and photonic functions, biological and medical species, etc. A facile one-step synthetic strategy to cross-linked polymeric nanoparticles (CLPN) is demonstrated via conventional radical copolymerization of phosphorous IL, 4-vinylbenzyl-triphenylphosphorous chloride, and ethylene dimethacrylate (EDMA) (Scheme 42).103 The catalytic performance of various quaternary phosphorous salts based catalysts for the cycloaddition reaction of CO2 to epichlorohydrin is investigated. Among all the catalysts, the one with the molar ratio of EDMA to PIL of 2:1 exhibits the highest catalytic activity (cyclic carbonate yield and selectivity >99%), which is presumably due to its nanostructure providing larger specific surface area and higher P content. CLPN could be reused for the cycloaddition reaction in five runs with only slight loss of its catalytic activity.
 | ||
| Scheme 42 Synthesis of cross-linked polymeric nano-particles based on PPIL-co-PEDMA.103 | ||
Tubular microporous organic networks bearing imidazolium salts (T-IM) can be prepared by Sonogashira coupling of tetrakis(4-ethynylphenyl)methane and diiodoimidazolium salts (Scheme 43), which show promising catalytic activities in heterogeneous conversion of CO2 into cyclic carbonates.104 The T-IM shows a maximum TOF up to 142 h−1 at 160 °C under 3 MPa CO2, and 130 h−1 at 150 °C and 1 MPa CO2. The recovered T-IM can give the nearly same reactivity as the original one.
 | ||
| Scheme 43 Preparation of porous organic networks bearing imidazolium salts.104 | ||
Synthesis of optically active cyclic carbonate using ionic liquids as co-catalysts
Direct synthesis of optically active cyclic carbonates from the racemic epoxides has been realized by Lu et al.105 based on the proposal that if an electrophile can selectively complex one enantiomer of racemic epoxides, the attack of nucleophile or activated CO2 to the coordinated epoxide on the less substituted carbon regioselectively would lead to enantioselective ring-opening of epoxide and then further form chiral cyclic carbonates via intramolecular cyclic elimination. A convenient route to optically active PC by a catalytic kinetic resolution process resulting from the coupling reaction of CO2 and racemic epoxides is developed using simple chiral SalenCo(III)/quaternary ammonium halide catalyst system. Solvent-free reaction of racemic PO (0.5 mol) with 0.55–0.60 equiv. of CO2 in the presence of 0.1 mol% of SalenCo(III)(O2CCCl3) complex 10b (Scheme 44) using as chiral electrophile in conjunction with 0.1 mol% of n-Bu4NBr as nucleophile proceeds within 2 h at room temperature to afford a mixture of unreacted epoxide and PC with moderate enantioselectivity (PC ee value: 50.5%). The formation rate of PC is up to a TOF of 245 h−1. The coexistence of a quaternary ammonium salt such as n-Bu4NBr could be essential to promote this reaction. The use of n-Bu4NCl is more beneficial for improving enantiomeric purity of the products (ee value: 70.2%), but pose a pronounced negative effect on the rate. | ||
| Scheme 44 Racemic propylene oxide/CO2 coupling results.105 | ||
Based on this report, various chiral catalysts for the reaction of CO2 with PO at atmospheric pressure have been developed to generate enantiomerically enriched PC. The best selectivity can be achieved with a CoIII(salen)-trifluoroacetyl complex and bis(triphenyl phosphoranylidene)ammonium fluoride (PPN+F−) as catalysts, affording PC in 40% yield and 83% ee value (Scheme 45).106
 | ||
| Scheme 45 CoIII(salen)-trifluoroacetyl/ PPN+F− catalytic system for enantiospecific addition of CO2 to propylene oxide.106 | ||
Chiral Co(salen) complexes bearing the Lewis acid of elemental group IIIA can efficiently catalyze the reactions of CO2 with epoxides in the presence of catalytic amounts of alkali metal salts, quaternary ammounium halide or ILs.107 For the asymmetric cycloaddition reaction of CO2 and PO catalyzed by Co(III) Salen-AlCl3/BMImOH (Scheme 46), the maximum ee value of PC (83.2%) can be achieved at 1:0.001:0.0001 PO/catalyst/cocatalyst molar ratio, 0.5 MPa CO2 pressure, 25 °C, 3 h, with 45.1% PC yield.
![Schematic diagram of chiral (salen)Co complexes and [BMIm]OH.107](/image/article/2011/RA/c1ra00307k/c1ra00307k-s46.gif) | ||
| Scheme 46 Schematic diagram of chiral (salen)Co complexes and [BMIm]OH.107 | ||
The catalyst system of chiral SalenCo(OAc)/chiral IL has been developed to catalyze the asymmetric cycloaddition reaction of CO2 and epoxides yielding the chiral cyclic carbonates (Scheme 47).108 The chiral ILs TBAX (X = amino acidic anions, tartaric acidic anions, lactic acidic anion) are used to be cocatalysts. The highest ee value of (S)-PC (74.6%) is achieved with [TBA][L-Pro] as cocatalyst. Various epoxides works well in this asymmetric cycloaddition in the presence of the catalyst SalenCo(OAc) /[TBA]2[L-Tar] at room temperature. Epichlorohydrin and 1,2-epoxybutane can furnish corresponding cyclic carbonates with higher ee values (21.3% and 47.2% respectively), and styrene oxide and 1,2-epoxy-3-phenoxy propane can acquire corresponding cyclic carbonates with lower ee values (3% and 4% respectively).
 | ||
| Scheme 47 Asymmetric cycloaddition of CO2 to epoxides.108 | ||
Dimethyl carbonate synthesis via a two-step process
Dimethyl carbonate (DMC) has been drawing much attention as a safe, non-corrosive and environmentally friendly building block for the production of polycarbonate and other chemicals,109 additive to fuel oil owing to a high octane number110 and electrolyte in lithium batteries due to its high dielectric constant.111 From the viewpoint of green chemistry, the two-step transesterification process utilizing CO2 as a raw material (Scheme 48) could be more attractive in comparison with other commercial processes.112,113 In the past decades, numerous catalysts have been proposed for the transterification of alkylene carbonate with methanol including ILs.114,115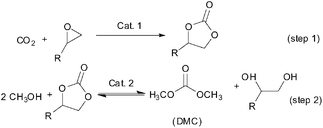 | ||
| Scheme 48 Two-step process for DMC production utilizing CO2 as a raw material. | ||
The synthesis of DMC from EC and methanol is carried out using ILs comprising cations of 1-ethyl-3-methylimidazolium (EMIm+), 1-butyl-3-methylimidazolium (BMIm+), 1-hexyl-3-methylimidazolium (HMIm+) and anions of Cl−, BF4−, PF6−. The order of the EC conversion decreases (from 76.3% to 67.0%) as the size of the cation of the imidazolium salt ILs increases from EMIm+ to HMIm+. The conversion of EC increases in the order of PF6− < BF4− < Cl− (from 56.3% to 76.3%), which is consistent with the order of the nucleophilicity of anions. A high pressure of CO2 is favorable for the inhibition of EC decomposition to ethylene oxide (EO) and CO2.116
We have synthesized a series of DABCO-based ILs to catalyze the transesterification of EC with methanol (Scheme 49), the cation of which has the potential to activate methanol by the tertiary nitrogen.114 Both DMC yield and selectivity decrease remarkably as the alkyl length of the cation increasing from C4 to C8, probably due to reducing hydrophilicity as well as solubility and thus decreasing reactivity of the IL in methanol.117 For the effect of anions, the catalytic efficiency increases in the order of Cl− < Br− < OH− (n = 3), BF4− < Br− < PF6− < Tf2N− (n = 7). 1-Butyl-4-azo-1-azoniabicyclo[2.2.2]octane hydroxide ([C4DABCO]OH) exhibits high activity and 81% DMC yield together with 90% EC conversion is obtained under mild reaction conditions (EC, 10 mmol; MeOH, 150 mmol; catalyst loading, 1 mol%; 70 °C; 6 h). Notably, the catalyst could be recycled four times without loss of catalytic activity.
 | ||
| Scheme 49 Lewis basic ionic liquid-catalyzed synthesis of DMC via transterification of EC with methanol.114 | ||
However, the major disadvantages of the process are high energy consumption and high investment due to the requirement of separation of the intermediate (cyclic carbonate). Therefore, it is very desirable to integrate the cycloaddition of CO2 with epoxide and the transterification of cyclic carbonate with methanol into a one-pot reaction. We have designed a binary catalyst (n-Bu4NBr/n-Bu3N), in which n-Bu4NBr would be active towards cycloaddition reaction (Scheme 48, step 1) and n-Bu3N could be effective for the transesterification process (Scheme 48, step 2).118 Under optimum reaction conditions (catalyst loading, 2.5 mol%; 150 °C; 15 MPa; 8 h), PO, SO and 2-(phenoxymethyl)oxirane exhibits significant activity with 59%, 80% and 84% of DMC yield, respectively. However, intermediate (cyclic carbonate) is the main product (50%), and the yield of DMC was merely 12% with incomplete conversion of epoxide (70%) when chloromethyl oxide is employed.
We have also developed a recyclable binary catalyst system, BrBu3PPEG6000PBu3Br and K2CO3/PEG6000 for DMC synthesis from PO, CO2 and methanol, which displays high activity for this process under mild reaction conditions, even under low CO2 pressure (2 atm).119 The catalyst (K2CO3/BrBu3PPEG6000PBu3Br) can be solidified by adding ether and cooling after the reaction, and recovered by a simple filtration, without catalyst leaching detected by 31P NMR. Synthesis of cyclic carbonate at 120 °C, 1 MPa, 6 h and then methanol is added, and the mixture is refluxed for 30 min at 100 °C, finally the reaction is continued for another 1 h while simultaneously removing the product DMC through distillation under atmospheric pressure. The yield of DMC can reach 97.5% in the fourth run of the catalyst.
Synthesis of urea derivatives and carbamates
Urea derivatives are an important class of carbonyl compounds and useful chemical intermediates in the synthesis of pharmaceuticals, agricultural chemicals and dyes; and they were also used as antioxidants in gasoline and additives in plastics.120 On the other hand, conventionally preparative methodologies of urea derivatives are based on the use of dangerous reagents such as phosgene and isocyanates.121 Nowadays, replacement of these hazardous reagents in chemical processes is one of the main goals in green chemistry. Therefore, the synthesis of ureas starting from CO2 has drawn much attention because CO2 is a renewable, abundant, cheap, and nontoxic source of functional carbon unit.122Deng and co-workers proposed an effective process for the direct synthesis and separation of symmetric urea derivatives in good yield from amines using CO2 as the carbonyl source catalyzed by a recyclable catalytic system consists of an IL and base (CsOH) (Scheme 50),24 to avoid the need for stoichiometric quantities of dehydrating agent. If the conversion can be sufficiently high, the desired product precipitates on adding about 10 mL water into the reaction mixture as urea is insoluble in water whereas CsOH and the IL are water-soluble. The solid product could be recovered by filtration. 98% yield of 1,3-dicyclohexylurea is obtained when BMImCl containing CsOH is employed (4 h). As to primary amine with a linear chain (n-hexylamine), a longer reaction time (6 h) is needed to obtain similar yield (6 h). Surprisingly, only 27% yield of urea can be achieved for the carbonylation of aromatic amine (aniline). BMImCl/CsOH could be recovered and reused after it is distilled to remove water, and a yield of 93% of 1,3-dicyclohexylurea can be obtained when CsOH/BMImCl system is used for a third time.
![Synthesis of urea derivatives catalyzed by [BMIm]Cl/CsOH.](/image/article/2011/RA/c1ra00307k/c1ra00307k-s50.gif) | ||
| Scheme 50 Synthesis of urea derivatives catalyzed by [BMIm]Cl/CsOH. | ||
The synthesis of disubstituted ureas from amines and CO2 using a basic IL 1-n-butyl-3-methyl imidazolium hydroxide ([BMIm]OH) as the catalyst has been also reported under solvent-free conditions without using any dehydrating regent (Scheme 51).25 At 170 °C, the yield of 1,3-dibutylurea can reach maximum, i.e. 55.1%, with [BMIm]OH loading as 15 mol%. The linear amines show better reactivity than branched amines. Benzylamine (urea yield: 46.1%) and cyclohexylamine (urea yield: 47.9%) give a similar activity. No product could be detected when aniline is used as the reactant. The possible reaction mechanism is depicted in Scheme 52. First, carbamate salt is formed from amine and CO2, and then the ion exchange reaction takes place between the carbamate salt and [BMIm]OH. As a result, the carbamate anion can be activated by the [BMIm]+ and the ammonium cation is reduced to amine in the presence of hydroxyl, along with the formation of H2O. Finally, regeneration of [BMIm]OH and formation of urea could complete the catalytic reaction cycle.
 | ||
| Scheme 51 Synthesis of N,N-dibutyl urea from n-butylamine and CO2.25 | ||

Recently, a catalyst system containing M(acac)x/BMMImCl (M = Co, Ni, Fe, Cu, Zn) for the carbonylation of amines and CO2 with 15%–25% aniline conversions and 87%–93% N,N′-diphenylurea (DPU) selectivities has been reported (Scheme 53).123 Among all the metal complexes, Co(acac)3 exhibits relatively higher catalytic activity, i.e., 25% conversion of aniline and 88% selectivity of DPU at 160 °C. The aromatic amines with an electron-donating group give higher conversion than those with an electron-withdrawing group. Aliphatic amines show much higher yields to the corresponding products (45%–81%). In addition, the Co(acac)3/BMMImCl catalyst system could be reused at least four times with a slight decrease in DPU yield.

An electrochemical procedure for the synthesis of organic carbamates from amines and CO2 can be also performed in IL.124 Selective cathodic reduction of CO2 successfully occurs in CO2-saturated IL [BMIm][BF4] solutions containing amines, followed by addition of EtI as an alkylating agent (Scheme 54).
 | ||
| Scheme 54 Electrochemical procedure for the synthesis of carbamates from amines and carbon dioxide.124 | ||
Synthesis of oxazolidinones
The [2+3] coupling reaction between CO2 and aziridines is one of the few commercial routes using CO2 as a raw material to afford the 5-membered materials, which are important heterocyclic compounds showing wide applications as intermediates and chiral auxiliaries in organic synthesis. Therefore, a growing effort has been devoted to developing efficient methodologies for producing oxazolidinones. From the viewpoint of green chemistry, the cycloaddition procedure utilizing CO2 as a feedstock is more attractive in comparison with those processes including carbonylation of amino alcohols with phosgene, CO, and reaction of propargylamine/ propargylic alcohol with CO2.20Selective synthesis of 2-oxazolidinone by cycloaddition of CO2 with aziridine is reported by employing tetraalkyl ammonium salts as the catalysts (Scheme 55).125 The reaction of 2-methylaziridine (MeAz) with CO2 is performed and 4-methyl-1,3-oxazolidin-2-one is given as a sole product. The catalyst efficiency largely depends on the counter anion and its order is found to be Br > I > Cl. The reaction catalyzed by TBAB can be performed at ambient temperature in the yield of 95% under atmospheric pressure using THF as the solvent.
 | ||
| Scheme 55 Cycloaddition of CO2 with 2-methylaziridine catalyzed by tetrabutylammonium halide.126 | ||
Recently, efficient synthesis of 2-oxazolidinone by cycloaddition of CO2 with aziridine was catalyzed by NH4I (Scheme 56).127 When the reaction is run at 0 °C for 4 h, the yield was 99%, with 96% of that material being the major isomer. In contrast, when the temperature of the reaction is increased from room temperature to 45 °C, the regioselectivity decreases to less than 80% of the major isomer.
 | ||
| Scheme 56 Cycloaddition of CO2 with aziridine catalyzed by NH4I.127 | ||
An easily recyclable catalyst, quaternary ammonium bromide covalently bound to PEG (PEG6000(NBu3Br)2) has been developed for the cycloaddition reaction of aziridines to CO2 under mild conditions without utilization of additional organic solvents or cocatalysts.128 As shown in Table 1, 5-aryl-2-oxazolidinones are obtained in high yield with excellent regioselectivity. The reactions of aziridines bearing alkyl groups at the nitrogen atom proceed smoothly and good yields are achieved within 20 min (Table 1, entries 1, 2, 4 and 5). 2-Phenylaziridine displays a relatively low selectivity due to the formation of self-oligomers (entry 3). The substrates bearing a branched alkyl group at the nitrogen atom shows slower reaction rate due to the steric interactions (entries 7–14). With regard to regioselectivity, if R1 is an aryl group, the intermediate corresponding to 12 would be more stable than 13 and thus 12 would be predominantly formed; in contrast, if R1 is an alkyl group, 13 would be favored, which in turn results in dominantly producing 13 (Scheme 57).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time | Conv.(%)b | Isolated yield (%)c | Regioselectivity (%)d |
| a Reaction condition: PEG6000(NBu3Br)2, 32.4 mg, 0.005 mmol; substrate, 2 mmol; CO2, 8 MPa; 100 °C. b Determined by GC. c The total yield of 12 and 13. d Molar ratio of 12 to 13. e 1,2,4,5-Tetraphenylpiperazine and 1,2,3,4-tetraphenylpiperazine were detected by LC-MS. | |||||
| 1 | 11a | 20 min | >99 | 95 | 92:8 |
| 2 | 11b | 15 min | >99 | 83 | 93:7 |
| 3 | 11c | 15 min | >99 | 59 | 86:14 |
| 4 | 11d | 20 min | 99 | 89 | 93:7 |
| 5 | 11e | 25 min | >99 | 94 | 91:9 |
| 6 | 11f | 45 min | >99 | >99 | 96:4 |
| 7 | 11g | 72 h | 50 | 49 | 100 |
| 8 | 11h | 24 h | >99 | 96 | 99:1 |
| 9 | 11i | 14 h | >99 | 93 | 96:4 |
| 10 | 11j | 12 h | >99 | 91 | 91:9 |
| 11 | 11k | 11 h | >99 | 88 | 94:6 |
| 12 | 11l | 10 h | 97 | 94 | 99:1 |
| 13 | 11m | 10 h | >99 | 94 | 99:1 |
| 14 | 11n | 15 h | >99 | 95 | 100 |
| 15 | 11o | 1 h | 99 | 88 | 20:80 |
| 16e | 11p | 24 h | 100 | ||

 | ||
| Scheme 57 A possible mechanism for PEG6000(NBu3Br)2-promoted cycloaddition reaction of CO2 with aziridines.128 | ||
Furthermore, the reaction of (S)-1-butyl-2-phenylaziridine (S-11e) with CO2 affords S-12e in 91.4% yield and S-13e in 8.6% yield with retention of stereochemistry (Scheme 58), further supporting the mechanism. Besides, the catalyst could be recovered by centrifugation and reused without significant loss of catalytic activity and selectivity.
 | ||
| Scheme 58 The cycloaddition of (S)-1-butyl-2-phenylaziridine to CO2.128 | ||
Our group has also developed a series of easily prepared Lewis basic ILs (Scheme 59) as recyclable and efficient catalysts for selective synthesis of 5-aryl-2-oxazolidinones from aziridines and CO2 without utilization of any organic solvent or additive.20 Both the yield and selectivity of 12a decrease markedly as the alkyl chain length of the cation increases from C4 to C12. Halide anions like Cl−, Br− give good results; whereas, Tf2N−, PF6− and BF4− are found to be inactive. For the catalyst recyclability, no significant drop in either the yield or selectivity of 12a is detected after four successive cycles. To gain a deeper insight into the reaction mechanism, in situ FT-IR spectroscopy under CO2 pressure is employed to identify the possible intermediates during the reaction. Interestingly, the absorption peak of the carbonyl group migrates from 1770 cm−1 (11a-CO2 or C4DABCO-CO2 carbamate salt) to 1740 cm−1 (oxazolidinone) when 11a is used as the substrate (Fig. 4), presumably implying the activation of CO2 by Lewis basic tertiary nitrogen atoms.
 | ||
| Scheme 59 Ionic liquids used in this study as catalysts for the synthesis of oxazolidinones.20 | ||
![Results of in situ IR spectroscopy under CO2 pressure monitoring at various reaction time (min). Reaction conditions: (A) triethylamine (5 mmol), 120 °C, CO2 (9 MPa); (B) 11a (5 mmol), 120 °C, CO2 (9 MPa); (C) 11a (5 mmol), [C4DABCO]Br (1 mmol), 90 °C, CO2 (6 MPa). 1780 and 1770 cm−1 correspond to peaks for carbonyl group of [Et3N–CO2], and [11a-CO2]/[C4DABCO-CO2] carbamic salt, respectively. 1740 cm−1 can be absorption of carbonyl group of oxazolidinones.20](/image/article/2011/RA/c1ra00307k/c1ra00307k-f4.gif) | ||
| Fig. 4 Results of in situ IR spectroscopy under CO2 pressure monitoring at various reaction time (min). Reaction conditions: (A) triethylamine (5 mmol), 120 °C, CO2 (9 MPa); (B) 11a (5 mmol), 120 °C, CO2 (9 MPa); (C) 11a (5 mmol), [C4DABCO]Br (1 mmol), 90 °C, CO2 (6 MPa). 1780 and 1770 cm−1 correspond to peaks for carbonyl group of [Et3N–CO2], and [11a-CO2]/[C4DABCO-CO2] carbamic salt, respectively. 1740 cm−1 can be absorption of carbonyl group of oxazolidinones.20 | ||
Reactions of propargylic alcohols, aliphatic primary amines, and CO2 are catalyzed by CuCl/[BMIm]BF4 system to produce the corresponding 5-methylene-1,3-oxazolidin-2-ones under relatively mild conditions (Scheme 60).129 Some other ILs, such as [BMIm]PF6 and N-butylpyridinium tetrafluoroborate ([BPy]BF4) also work well. Reactions of various commercially available propargylic alcohols and amines with CO2 have been successfully performed under the optimum reactions conditions ([BMIm]BF4, 3.0 mL; CuCl, 0.2 mmol; amine, 10 mmol; propargylic alcohol, 10 mmol; CO2 pressure, 2.5 MPa, 100 °C). An aliphatic diamine, hexamethylenediamine, could also undergo the three-component reaction to give the double nucleus compound in 82% isolated yield.
 | ||
| Scheme 60 General reaction of aliphatic primary amines, and CO2.129 | ||
Hydrogenation of CO2 promoted by ionic liquids
The hydrogenation of CO2 to produce formic acid is an attractive reaction that has been carried out in organic solvents,130 water,131 and scCO2.132 Han et al. have reported the first use of a TSIL, that has a tertiary amino group (N(CH3)2) on the cation, 1-(N,N-dimethylaminoethyl)-2,3-dimethylimidazolium trifluoromethane sulfonate ([MAMMIm] OTf) (Scheme 61), as a base that promotes the hydrogenation of CO2 to form formic acid.23 Ruthenium immobilized on silica (“Si”–(CH2)3NH(CSCH3)–RuCl3–PPh3) is used as a heterogeneous catalyst, which can be dispersed in the IL aqueous solution during the reaction. The IL is used to shift the reaction equilibrium and therefore it is easier for the IL to form a salt with formic acid at higher concentrations. The unique feature of this approach is that the formic acid can be recovered easily and both the IL and catalyst can be reused after a simple separation process (Fig. 5). No evident decrease of TOF is observed after recycling the catalyst and IL for five times.![Synthesis of the ionic liquid [MAMMIm][TfO].23](/image/article/2011/RA/c1ra00307k/c1ra00307k-s61.gif) | ||
| Scheme 61 Synthesis of the ionic liquid [MAMMIm][TfO].23 | ||
 | ||
| Fig. 5 Hydrogenation reaction and recovery of the product, catalyst, and ionic liquid. Reprinted with permission from ref. 23. Copyright 2008 John Wiley and Sons. | ||
They also reported a similar TSILs 1,3-di(N,N-dimethylaminoethyl)-2-methylimidazolium trifluoromethane sulfonate ([DAMIm][TfO]) (Scheme 62), which has two tertiary amino groups on the cation, in the use of CO2 hydrogenation.133 The molar ratio of formic acid/IL at 60 °C and 18 MPa reaches about 1.76 with increasing reaction time up to 8 h. After this time, the ratio increases slowly and finally reaches a constant value of around 2 at 12–14 h. The TOF value increases with the amount of water added, and reaches a value of 109 h−1 when 1.0 g water is added.
![Synthetic route133 to the IL [DAMIm][TfO]. Reaction conditions: a) BrCH2CH2NH2·HBr, acetonitrile, 78 °C, 24 h; b) NaOH, methanol, 2 h; c) BrCH2CH2NH2·HBr, acetonitrile, 78 °C, 10 h; d) HCHO, HCOOH, 100 °C, 36 h; combined yield of 30% for steps a–d; e) NaOH, F3CSO3Na, methanol, 97%.](/image/article/2011/RA/c1ra00307k/c1ra00307k-s62.gif) | ||
| Scheme 62 Synthetic route133 to the IL [DAMIm][TfO]. Reaction conditions: a) BrCH2CH2NH2·HBr, acetonitrile, 78 °C, 24 h; b) NaOH, methanol, 2 h; c) BrCH2CH2NH2·HBr, acetonitrile, 78 °C, 10 h; d) HCHO, HCOOH, 100 °C, 36 h; combined yield of 30% for steps a–d; e) NaOH, F3CSO3Na, methanol, 97%. | ||
Baker and Tumas et al. reported the hydrogenation of CO2 in the presence of dialkylamines to produce N,N-dialkylformamide through polar intermediates which would be soluble in the IL phase and not in scCO2 (Scheme 63).134 Carbamate could be completely converted to DMF after 4 h at 80 °C using 5.5MPa hydrogen under a total pressure of 27.6 MPa.
 | ||
| Scheme 63 Hydrogenation of carbon dioxide in the presence of dialkylamines to produce N,N-dialkylformamides.134 | ||
Conclusions and perspective
CO2 as an abundant, typical renewable C1 source as well as an important “greenhouse” gas has been drawing more and more attention. And chemists may often think “what we can do with CO2 from a scientific and engineering point of view?” Obviously, CO2 capture and storage looks particularly effective for temporarily preserving large volumes of CO2. In general, potential uses of CO2 as a raw material, as a sound green solvent, or as a Lewis acid catalyst or tunable reagent could offer profound advantages for both green chemistry and improved economics in terms of creating novel chemistry, reaction control, product separation, and operation simplification.As a renewable feedstock, the transformation of CO2 into useful chemicals would be particularly important and deserves worldwide attention. However, its inherent thermodynamic stability and kinetic inertness hinder the development of metal catalysts that achieve CO2 activation and functionalization. Based on the weak activation between CO2 and catalyst, various value-added chemicals such as carbonates and oxazolidinones have been synthesized using CO2 as a feedstock. The key point is the simultaneous activation of CO2 and the reactants such as epoxides and aziridines by the bifunctional catalysts. Based on this principle, dual-functional one-component catalysts have been designed, such as amino acid-based ILs, quaternary ammonium or quaternary phosphonium functionalized PEG and quaternary phosphonium functionalized transition metal salen complexes, to catalyze the reaction under mild conditions through the synergistic activation. In addition, homogeneous catalyst recovery and recycling could be achieved according to the characteristics of ILs/scCO2 or PEG/scCO2 two-phase system.
ILs (quaternary ammonium salts, imidazolium salts, etc.) have been found to be efficient catalysts for the cycloaddition reaction of epoxides and CO2, but catalyst separation from products are difficult. Silica-supported imidazolium ILs and the corresponding homogeneous catalysts give similar activity under supercritical conditions, with 96% yield and 99% selectivity of product. Notably, the silica-supported ammonium catalyst could show better performance in facilitating cycloaddition reaction of CO2 with epoxide than the corresponding homogeneous counterpart, presumably due to dual activity of both reactants by hydroxyl group on the silica surface. The heterogeneous catalysts can be separated by simple filtration and the purity of the product can be up to 99% without further purification.85,86,94 Therefore, easily recoverable and highly efficient immobilized ILs catalysts have great potential to be used in continuous industrial production applications.
ILs, in particular TSILs and supported ILs have wide applications in chemical conversion of CO2, including the following aspects: synthesis of cyclic carbonates (including optically active cyclic carbonates) from cycloaddition reaction of CO2 and epoxides; dimethyl carbonate synthesis via a two-step process; synthesis of urea derivatives and carbamates through carbonylation reaction between CO2 and amines, synthesis of oxazolidinones from CO2 and aziridines and synthesis of formic acid via hydrogenation of CO2. Moreover, TSILs including amino-functionalized, hydroxyl-functionalized or PILs et al. are efficient absorbents for chemical absorption of CO2, owing to the merits of negligible vapor pressures, high thermal stabilities, tunable properties and so on. Although significant advances have been made, there remains the challenge to employ ILs as CO2 absorbents for designing industrial processes which are environmentally friendly and economically feasible since ILs could commonly be toxic and expensive; in particular, their synthesis usually requires multiple synthetic steps and thus limits their commercial viability. In order to become alternative candidates for industrial CO2 capture, ILs with characteristics of high capacity, low viscosity and economy still need to be developed.
Carbon dioxide sequestration is a technology that is being explored to curb the anthropogenic emission of CO2 into the atmosphere. With the environmental issues and energy crisis problem associated with “greenhouse gas effect” being more and more prominent, CO2 capture-fixation-conversion has been a worldwide hot topic for sustainable development. Given that CO2 as a starting material involving in the reported reactions is pure CO2 as raw material, it is important to utilize CO2 in industrial emissions. This requires achievement of high efficient fixation of CO2 in industrial emissions first of all and then conversion of the captured CO2. It will have great significance to capture CO2 and convert into useful chemical products or intermediates simultaneously, taking into account the atom economy and other green issues during the waste disposal process. Turning CO2 into a feedstock for producing useful commodity chemicals under mild conditions would indeed circumvent most of drawbacks of the energy-consumed steps of CO2 desorption, absorbent regeneration as well as of CO2 transportation and disposal in geological cavities, oceans or elsewhere.
In summary, numerous strategies have so far been proposed for chemical absorption of CO2. Although significant advances have been made, there are still intrinsic drawbacks such as extensive energy consumption for CO2 desorption, low capture efficiency and slow sorption kinetics to be addressed. In particular, extensive energy input in desorption process would be a crucial barrier to realizing practical CCS. Hence, reducing the energy requirement is an essential prerequisite for a breakthrough in absorption techniques.
On the other hand, the reactions involving CO2 are commonly carried out at high pressure, which may not be economically suitable and also pose safety concerns. The challenge is to develop catalysts that are capable of activating CO2 under low pressure (preferably at 1 atm), and thus incorporating CO2 into organic molecules catalytically. In this context, efforts to convert CO2 to useful chemicals under mild conditions will inevitably rely on its activation.
We proposed an alternative concept (CCU) to addressing the energy penalty problem in the CCS process. The essence of our strategy is to use task-specific ILs for CO2 capture and whereby substantial activation, which renders this system suitable for accomplishing chemical transformation of CO2 under low pressure (ideally at 1 atm), getting rid of the desorption step. It is of interest to note that the ammonium alkylcarbonate salt formed upon CO2 uptake could result in CO2 activation, which was confirmed by NMR, in situ FT-IR and showed excellent reactivity in incorporating captured CO2 into organic molecules catalytically under low pressure.
Subsequent reaction135 can be performed by using the captured CO2 as a starting material and the absorbent as a catalyst to validate our strategy. For example, the liquid amidinium carbonate salt formed upon CO2 (gas, 1 atm) absorption with the IL directly reacts with n-butylamine successfully to afford the target product i.e. dibutyl urea in almost quantitative yield (96%) of 1,3-dibutylurea without the use of additives. It is worth mentioning that CO2 absorption could lead to its activation, almost no additional energy input is required for desorption process with this strategy. Overall, the fixed CO2 could be catalytically converted to value-added chemicals under extremely mild reaction condition (1 atm, 40 °C and metal-free process). We believe this protocol will find abundant applications in CO2 chemistry, in particular, carbon capture and chemical utilization of CO2 in industry.
Great efforts have been directed towards constructing C–C, C–O and C–N bond on the basis of CO2 activation for production of a wide set of value-added organic compounds like solvents, fuels, fine/bulk chemicals, pharmaceuticals and polymers. There remain many opportunities to be explored further in CO2 chemistry on the topic of CO2 in organic synthesis, particularly in terms of functionalization of CO2 through its activation by using active catalysts like ILs, and on smartly utilizing dense CO2 as an environmentally benign solvent, or using CO2 as a catalyst or tunable reagent. It can be predicted that functionalized, low toxicity, made from readily available raw materials and easily prepared ILs will have bright prospects in the field of CO2 absorption, conversion and utilization. Technologies for CO2 fixation and conversion based on ILs will be transformed into productivity and promote the development of green chemistry and sustainable chemical processes.
Abbreviations
Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant Nos. 20672054, 20872073), and Research Fellowship for International Young Scientists from NSFC (21150110105), and the Committee of Science and Technology of Tianjin for financial support.References
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS
.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 2739–2751 CrossRef CAS
- J.-L. Wang, C.-X. Miao, X.-Y. Dou, J. Gao and L.-N. He, Curr. Org. Chem., 2011, 15, 621–646 CrossRef CAS
- L.-N. He, J.-Q. Wang and J.-L. Wang, Pure Appl. Chem., 2009, 81, 2069–2080 CrossRef CAS
-
L.-N. He, Z.-Z. Yang , A.-H. Liu and J. Gao, in Advances in CO2 Conversion and Utilization, American Chemical Society, 2010, vol. 1056, pp. 77–101 Search PubMed
- N. McCann, M. Maeder and M. Attalla, Ind. Eng. Chem. Res., 2008, 47, 2002–2009 CrossRef CAS
- J. Alejandre, J. L. Rivera, M. A. Mora and V. de la Garza, J. Phys. Chem. B, 2000, 104, 1332–1337 CrossRef CAS
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS
- C. Wang, H. Luo, D. e. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CAS
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC
- S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC
- Z.-Z. Yang, L.-N. He, S.-Y. Peng and A.-H. Liu, Green Chem., 2010, 12, 1850–1854 RSC
- Z. Z. Yang, L. N. He, C. X. Miao and S. Chanfreau, Adv. Synth. Catal., 2010, 352, 2233–2240 CrossRef CAS
- Y. Gu, F. Shi and Y. Deng, J. Org. Chem., 2004, 69, 391–394 CrossRef CAS
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS
- F. Shi, Y. Deng, T. SiMa, J. Peng, Y. Gu and B. Qiao, Angew. Chem., Int. Ed., 2003, 42, 3257–3260 CrossRef
- T. Jiang, X. Ma, Y. Zhou, S. Liang, J. Zhang and B. Han, Green Chem., 2008, 10, 465–469 RSC
- F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS
- F. Li, L. Xiao, C. Xia and B. Hu, Tetrahedron Lett., 2004, 45, 8307–8310 CrossRef CAS
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 403–408 RSC
- M. M. Dharman, H.-J. Choi, D.-W. Kim and D.-W. Park, Catal. Today, 2011, 164, 544–547 CrossRef CAS
- F. Barzagli, F. Mani and M. Peruzzini, Green Chem., 2011, 13, 1267–1274 RSC
- E. J. Beckman and P. Munshi, Green Chem., 2011, 13, 376–383 RSC
- Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS
- J. Ranke, S. Stolte, R. Störmann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS
- T.P.T. Pham, C. W. Cho and Y. S. Yun, Water Res., 2010, 44, 352–372 CrossRef
- J. S. Torrecilla, J. Palomar, J. Lemus and F. Rodriguez, Green Chem., 2010, 12, 123–134 RSC
- M. Alvarez-Guerra and A. Irabien, Green Chem., 2011, 13, 1507–1516 RSC
- S. Zhang, Y. Chen, F. Li, X. Lu, W. Dai and R. Mori, Catal. Today, 2006, 115, 61–69 CrossRef CAS
- H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357–358, 97–102 CrossRef CAS
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS
- Á. Pérez-Salado Kamps, D. Tuma, J. Xia and G. Maurer, J. Chem. Eng. Data, 2003, 48, 746–749 CrossRef CAS
- M. B. Shiflett and A. Yokozeki, J. Phys. Chem. B, 2007, 111, 2070–2074 CrossRef CAS
- S. Raeissi and C. J. Peters, J. Chem. Eng. Data, 2008, 54, 382–386
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Chem.–Eur. J., 2006, 12, 4021–4026 CrossRef CAS
- G. Yu, S. Zhang, X. Yao, J. Zhang, K. Dong, W. Dai and R. Mori, Ind. Eng. Chem. Res., 2006, 45, 2875–2880 CrossRef CAS
- Y. Zhang, S. Zhang, X. Lu, Q. Zhou, W. Fan and X. Zhang, Chem.–Eur. J., 2009, 15, 3003–3011 CrossRef CAS
- Y.-Y. Jiang, G.-N. Wang, Z. Zhou, Y.-T. Wu, J. Geng and Z.-B. Zhang, Chem. Commun., 2008, 505–507 RSC
- H. Yu, Y.-T. Wu, Y.-Y. Jiang, Z. Zhou and Z.-B. Zhang, New J. Chem., 2009, 33, 2385–2390 RSC
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879–884 RSC
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis, Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Chem.–Eur. J., 2009, 15, 7619–7627 CrossRef CAS
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102–1102 CrossRef CAS
- L. Phan, D. Chiu, D. J. Heldebrant, H. Huttenhower, E. John, X. Li, P. Pollet, R. Wang, C. A. Eckert, C. L. Liotta and P. G. Jessop, Ind. Eng. Chem. Res., 2008, 47, 539–545 CrossRef CAS
- Y. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 RSC
- C. Wang, S. M. Mahurin, H. Luo, G. A. Baker, H. Li and S. Dai, Green Chem., 2010, 12, 870–874 RSC
- C. Wang, H. Luo, X. Luo, H. Li and S. Dai, Green Chem., 2010, 12, 2019–2023 RSC
- C. Wang, X. Luo, H. Luo, D.-e. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Acc. Chem. Res., 2007, 40, 1208–1216 CrossRef CAS
- T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS
- D. J. Heldebrant, P. K. Koech, M. T. C. Ang, C. Liang, J. E. Rainbolt, C. R. Yonker and P. G. Jessop, Green Chem., 2010, 12, 713–721 RSC
- D. Camper, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 8496–8498 CrossRef CAS
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC
- A. B. Foster and J. M. Webber, Adv. Carbohydr. Chem., 1960, 15, 371–393 CAS
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef CAS
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC
- H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897 RSC
- H. S. Kim, J. J. Kim, H. Kim and H. G. Jang, J. Catal., 2003, 220, 44–46 CrossRef CAS
- J. Palgunadi, O. S. Kwon, H. Lee, J. Y. Bae, B. S. Ahn, N.-Y. Min and H. S. Kim, Catal. Today, 2004, 98, 511–514 CrossRef CAS
- J. Sun, S.-i. Fujita, F. Zhao and M. Arai, Green Chem., 2004, 6, 613–616 RSC
- J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS
- H. Xie, H. Duan, S. Li and S. Zhang, New J. Chem., 2005, 29, 1199–1203 RSC
- H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30–34 CrossRef CAS
- Y. J. Kim and R. S. Varma, J. Org. Chem., 2005, 70, 7882–7891 CrossRef CAS
- Y. Zhou, S. Hu, X. Ma, S. Liang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS
- H. Yang, Y. Gu, Y. Deng and F. Shi, Chem. Commun., 2002, 274–275 RSC
- K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876–1880 RSC
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS
- J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167–172
- T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664–1666 RSC
- S. Udayakumar, S.-W. Park, D.-W. Park and B.-S. Choi, Catal. Commun., 2008, 9, 1563–1570 CrossRef CAS
- H.-L. Shim, S. Udayakumar, J.-I. Yu, I. Kim and D.-W. Park, Catal. Today, 2009, 148, 350–354 CrossRef
- S. Udayakumar, M.-K. Lee, H.-L. Shim, S.-W. Park and D.-W. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS
- S. Udayakumar, V. Raman, H.-L. Shim and D.-W. Park, Appl. Catal., A, 2009, 368, 97–104 CrossRef CAS
- S. Udayakumar, M.-K. Lee, H.-L. Shim and D.-W. Park, Appl. Catal., A, 2009, 365, 88–95 CrossRef CAS
- K. Suzawa, M. Ueno, A. E. H. Wheatley and Y. Kondo, Chem. Commun., 2006, 4850–4852 RSC
- Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518–523 RSC
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS
- J. He, T. Wu, Z. Zhang, K. Ding, B. Han, Y. Xie, T. Jiang and Z. Liu, Chem.–Eur. J., 2007, 13, 6992–6997 CrossRef CAS
- Y. Zhao, J.-S. Tian, X.-H. Qi, Z.-N. Han, Y.-Y. Zhuang and L.-N. He, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC
- Y. Du, J.-Q. Wang, J.-Y. Chen, F. Cai, J.-S. Tian, D.-L. Kong and L.-N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS
- X. Dou, J. Wang, Y. Du, E. Wang and L. He, Synlett, 2007, 18, 3058–3062
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC
- Y. Xie, K. Ding, Z. Liu, J. Li, G. An, R. Tao, Z. Sun and Z. Yang, Chem.–Eur. J., 2010, 16, 6687–6692 CAS
- Y. Xiong, H. Wang, R. Wang, Y. Yan, B. Zheng and Y. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC
- H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917–919 RSC
- X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS
- A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401–4404 CrossRef CAS
- S.-W. Chen, R. B. Kawthekar and G.-J. Kim, Tetrahedron Lett., 2007, 48, 297–300 CrossRef CAS
- S. Zhang, Y. Huang, H. Jing, W. Yao and P. Yan, Green Chem., 2009, 11, 935–938 RSC
- S.-i. Fujita, B. M. Bhanage, Y. Ikushima and M. Arai, Green Chem., 2001, 3, 87–91 RSC
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS
- T. Wei, M. Wang, W. Wei, Y. Sun and B. Zhong, Green Chem., 2003, 5, 343–346 RSC
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475–494 CrossRef CAS
- Z.-Z. Yang, L.-N. He, X.-Y. Dou and S. Chanfreau, Tetrahedron Lett., 2010, 51, 2931–2934 CrossRef CAS
- H.-Y. Ju, M. Manju, D.-W. Park, Y. Choe and S.-W. Park, React. Kinet. Catal. Lett., 2007, 90, 3–9 CrossRef CAS
- B. M. Bhanage, S.-i. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC
- J.-S. Tian, J.-Q. Wang, J.-Y. Chen, J.-G. Fan, F. Cai and L.-N. He, Appl. Catal., A, 2006, 301, 215–221 CrossRef CAS
- J.-S. Tian, C.-X. Miao, J.-Q. Wang, F. Cai, Y. Du, Y. Zhao and L.-N. He, Green Chem., 2007, 9, 566–571 RSC
- B. Gabriele, G. Salerno, R. Mancuso and M. Costa, J. Org. Chem., 2004, 69, 4741–4750 CrossRef
- J. S. Nowick, N. A. Powell, T. M. Nguyen and G. Noronha, J. Org. Chem., 1992, 57, 7364–7366 CrossRef CAS
- D.-L. Kong, L.-N. He and J.-Q. Wang, Synlett, 2010, 1276–1280 CAS
- J. Li, X. Guo, L. Wang, X. Ma, Q. Zhang, F. Shi and Y. Deng, Sci. China Chem., 2010, 53, 1534–1540 Search PubMed
- M. Feroci, M. Orsini, L. Rossi, G. Sotgiu and A. Inesi, J. Org. Chem., 2006, 72, 200–203
- A. Sudo, Y. Morioka, E. Koizumi, F. Sanda and T. Endo, Tetrahedron Lett., 2003, 44, 7889–7891 CrossRef CAS
- A. Sudo, Y. Morioka, E. Koizumi, F. Sanda and T. Endo, Tetrahedron Lett., 2003, 44, 7889–7891 CrossRef CAS
- C. Phung and A. R. Pinhas, Tetrahedron Lett., 2010, 51, 4552–4554 CrossRef CAS
- Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef CAS
- Y. Gu, Q. Zhang, Z. Duan, J. Zhang, S. Zhang and Y. Deng, J. Org. Chem., 2005, 70, 7376–7380 CrossRef CAS
- R. Fornika, H. Gorls, B. Seemann and W. Leitner, J. Chem. Soc., Chem. Commun., 1995, 1479–1481 RSC
- J. Elek, L. Nádasdi, G. Papp, G. Laurenczy and F. Joó, Appl. Catal., A, 2003, 255, 59–67 CrossRef CAS
- P. Munshi, A. D. Main, J. C. Linehan, C.-C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963–7971 CrossRef
- Z. Zhang, S. Hu, J. Song, W. Li, G. Yang and B. Han, ChemSusChem, 2009, 2, 234–238 CrossRef CAS
- F. Liu, M. B. Abrams, R. T. Baker and W. Tumas, Chem. Commun., 2001, 433–434 RSC
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, Energy Environ. Sci., 2011 10.1039/c1ee02156g
| This journal is © The Royal Society of Chemistry 2011 |