New insights into the photo-tautomerisation process in β-carboline derivatives revealed by NMR spectroscopy
D.
Reyman
*a,
C.
Díaz-Oliva
a,
F.
Hallwass
b and
S. M.
Gonçalves de Barros
b
aDepartamento de Química Física Aplicada, Universidad Autónoma de Madrid, Cantoblanco, E-28049, Madrid, Spain. E-mail: dolores.reyman@uam.es
bDepartamento de Química Fundamental, Universidade Federal de Pernambuco, 50670-901, Recife, PE, Brazil
First published on 6th September 2011
Abstract
In this work, we present a spectroscopic study of the interactions of harmine (HI), 2-methylharmine (2MHI) and harmaline (HLINE) with acetic acid (AcOH) in low polarity media, such as 1,4-dioxane and chloroform. UV-Vis absorption, fluorescence spectroscopy and 1H NMR measurements show that, in all cases, a complex between these β-carboline derivatives (BCD) and acetic acid is formed. In addition, this complex exists in two different tautomeric forms in solution, depending on the concentration of acetic acid. The NMR data also indicates that electronic charge transfer takes place differently, depending on which tautomer (T1 or T2) is the most stable. Thus, when T1 is the most stable tautomer species, charge transfer induced by hydrogen bond interactions takes place from the pyrrole to the pyridine ring; however, for T2, the charge transfer occurs in reverse. We have also shown that loss of aromaticity in the pyridine ring strongly increases the acidity of the NH group of the pyrrolic ring of these derivatives.
I. Introduction
Prototropic tautomerism is an important class of isomerism in biological systems, that is essential for many biochemical processes,1 and may play a key role in DNA mutagenesis during replication. As such, numerous experimental and theoretical investigations have been conducted, in an attempt to explain the physicochemical changes that occur in DNA, which are responsible for generating mutations during the DNA replication process. However, despite these efforts, the mechanisms governing DNA mutagenesis are not clearly understood.2–22Tautomerism involves an equilibrium between two or more independent isomers (tautomers), with different positions of atoms or groups. Interconversion between these thermodynamically stable tautomers is possible, and often depends on different parameters, such as: temperature, light and solvent conditions (e.g. the acidic/basic properties of the medium surrounding the molecules). In fact, even small environmental variations may cause dramatic changes in the direction of this equilibrium, and consequently, in the biological properties of tautomeric systems,23–28 such as those responsible for DNA mutagenesis.
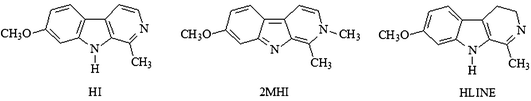
A special class of prototropic tautomeric systems is formed by N–heteroaromatic compounds which possess two or more nitrogen atoms in their structure. This class of tautomers, which include β-carboline (9H-pyrido[3,4-b]indole) derivatives (BCD),29–36 is of particular interest, both because of their ubiquitous nature and the fact that they comprise the basic elements of most of biological macromolecules. Structurally, these N–heteroaromatic molecules can be viewed as the condensation products of simpler individual N–heteroaromatic rings: a π-deficient pyridine ring fused to a π-excessive indole ring (see Scheme 1). The acidic/basic properties of the nitrogen atoms in these condensed molecules can vary greatly upon excitation; hence, the tautomerisation equilibrium position in S1 differs from that in S0.
 | ||
| Scheme 1 | ||
BCD have many interesting biological and photophysical properties, as well as biological activities, such as the ability to intercalate into DNA.37,38 In addition, BCD serve as an inhibitor of various enzymes such as: cyclin-dependent kinases (CDKs);39 topisomerase;40,41 and monoamine oxidase;42,43 and displays a broad spectrum of pharmacological properties, including: anxiolytic, hypnotic, anticonvulsant,44–46 antiviral,47 and antimicrobial activities.48
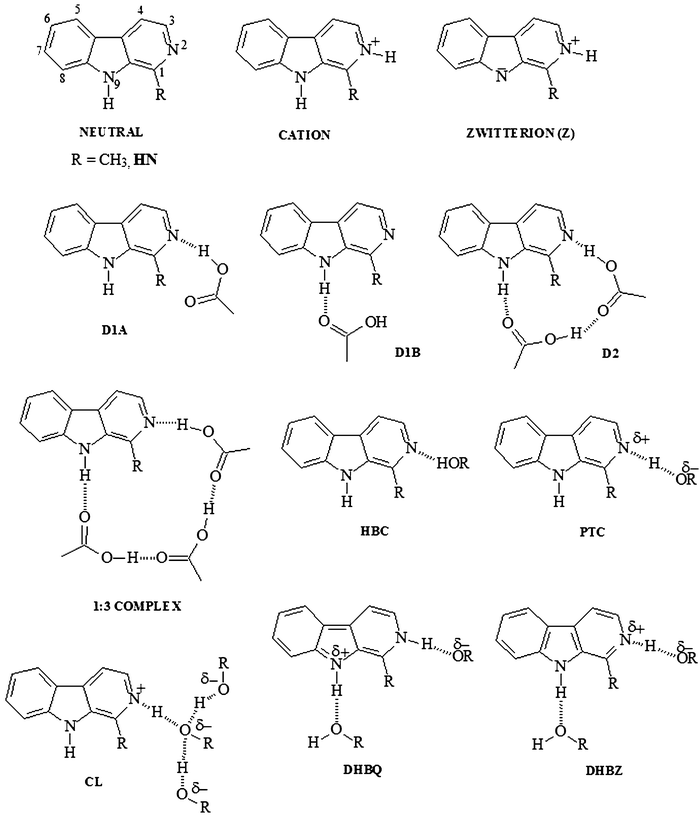
These derivatives display interesting photophysical properties that are associated with photo-induced changes in their electronic distribution, resulting in modification of the acidic and basic properties of the two nitrogen atoms at the β-carboline ring, and leading to variations in its reactivity. Upon excitation to the S1 state, the pyridinic nitrogen (Npd) becomes more basic, while the pyrrolic group (Npr–H) becomes more acidic. These changes may then promote a double proton transfer, when both hydrogen bonding donors and hydrogen bonding acceptors are present in the chemical environment surrounding the molecule. This double proton transfer induces a photo-tautomerisation process, thus complicating the photophysicochemistry of these derivatives.49,50Scheme 1 summarises some of the species proposed to explain this complex photophysicochemistry.51–57
Our knowledge of the mechanisms behind the formation of these species derives from research on the spectroscopic properties of different methylated BCD. However, controversy remains surrounding the structure, kinetics and formation mechanism of a species with an emission band at around 520 nm. Initially, this emission was observed in aqueous solutions of the β-carboline (BC) (or norharmane), at pH values greater than 12. A zwitterionic structure (Z, Scheme 2) was associated with this emission, in which the pyridinic and pyrrolic nitrogen atoms are protonated and deprotonated, respectively. Thus, the stability of this compound was only expected to be stable in highly polar media, and formation of this species was hypothesised to require the presence of both hydrogen-bonding donors and acceptors in the surrounding medium. However, this emission was observed experimentally to be more prevalent in low polarity solvents (e.g.dioxane, dichloromethane, chloroform, benzene and cyclohexane), in the presence of only a single hydrogen donor, such as 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP), or a single hydrogen bonding donor–acceptor, such as acetic acid (AcOH).
 | ||
| Scheme 2 | ||
In a study of the photophysics of N2-methylcarboline, which can be considered to be the prototype of β-carboline phototautomer, Carmona et al.54,58 hypothesised that the zwitterionic structure (Z) might also present a quinoid structure (Q), (Scheme 3). Furthermore, from the results of this study, two different ground state isomers54,58 were proposed: one with a quinoid structure (DHBQ) and another with a dipolar zwitterionic structure (DHBZ) (see Scheme 2). In fact, upon excitation, each isomer simultaneously gives rise to two emissions: one from a locally excited state (LE) at 360 nm, and another from a relaxed intramolecular charge transfer excited state (ICT), at 520 nm. The LE states of DHBQ and DHBZ isomers have ππ* and nπ* character, respectively. For the quinoid form, the photo-induced charge transfer process proceeds from the pyridinic to the pyrrolic nitrogen atoms; whereas for the zwitterionic form, the charge transfer process operates in the opposite direction. The simultaneous presence of partially charged donor and acceptor nitrogen atoms in these complexes facilitates the charge transfer process between these centres. Thus, they can acquire a quinoid (DHBQ), or zwitterionic (DHBZ) type of structure, depending on the polarity of the media.
 | ||
| Scheme 3 | ||
However, we believe that the quinoid and dipolar zwitterionic forms proposed by Carmona et al. are not isomers, but rather mesomeric structures originating from a single emission band (at approximately 520 nm). Accordingly, the emission maxima at 360 and 520 nm can be associated with a tautomeric equilibrium between species T1 (Npd, Npr–H, 360 nm) and T2 (Npd–H, Npr, 520 nm) (Scheme 4).
 | ||
| Scheme 4 | ||
Although T2 is only observed in S1, we assume that these hydrogen bonding interactions are also weakly present in the ground state, and become stronger in the S1 state, due to photo-induced changes in the electronic distribution mentioned above. This hypothesis was confirmed in recent research using 1H NMR spectroscopy.56 In this work, the spectra of 1-methyl-9H-pyrido[3,4-b]indole (or harmane, HN), (see Scheme 1) in different chloroform/AcOH mixtures was obtained. The H3, H4 and H5 hydrogens display greater variability, and shift to lower frequencies with increasing AcOH concentration. These chemical shift changes could be due to increased shielding, caused by hydrogen bond formation between the hydrogen atom of AcOH and the pyridinic nitrogen atom of HN. Hence, formation of a hydrogen bond (Npd⋯H–O), coupled with charge density delocalisation from the pyrrolic ring toward the pyridinic ring, shields the hydrogen atoms of this pyridine ring. The presence of this hydrogen bond is supported experimentally by a new signal that appears upon addition of AcOH. This signal is delocalised downfield with increasing AcOH concentration. At the same time, the Npr–H signal shifts to higher frequency with increasing AcOH concentration. The same effects can be produced by lowering the temperature at which spectra are recorded.56 Based on the above, the following mechanism was proposed to explain the observed emission maximum at approximately 520 nm in fluorescence spectra. A transformation of the type  is concomitant with
is concomitant with  . Hence, an increase in AcOH concentration drives a gradual transformation of the molecular hydrogen-bonded complex (T1) to a zwitterionic complex (T2 (Z)), which resonates with the quinoid structure (T2 (Q)) (see Scheme 4).
. Hence, an increase in AcOH concentration drives a gradual transformation of the molecular hydrogen-bonded complex (T1) to a zwitterionic complex (T2 (Z)), which resonates with the quinoid structure (T2 (Q)) (see Scheme 4).
The aim of the present study was to confirm these proposed mechanisms, and to explain the tautomeric equilibrium associated with these systems, by conducting spectroscopic studies on the interactions of three BCD with a hydrogen bonding acceptor/donor such as acetic acid, in low polarity solvents, such as dioxane and chloroform. Harmine (HI) and 2-methylharmine (2MHI) were chosen as models of T1 and T2 tautomers, respectively; while harmaline (HLINE) was chosen to study the effects of aromaticity on the tautomerization process (Scheme 1).
II. Experimental
All solvents used in the present study were of the highest available purity, and were purchased from Merck in Uvasol, or a similar grade. CDCl3 (99.9%), was purchased from Aldrich. HI, HLINE and 2MHI were purchased from Sigma.Absorption spectra were obtained using an HP 8453 UV-Vis spectrophotometer (Hewlett-Packard). Steady-state fluorescence measurements were performed on a Shimadzu RF-5301 PC spectrofluorimeter, with a 3 nm excitation bandwidth and a 1.5 nm emission bandwidth.
NMR measurements were performed in a 5 mm tube, in CDCl3 solution, on a Bruker Avance DRX 500, operating at 500.13 MHz for 1H resonances. Sample temperature was kept constant at 298 K. Chemical shifts were measured in reference to the residual CHCl3 solvent signal (at 7.262 ppm). The acquisition parameters of each spectrum consisted of 32 scans of 32 K data points, with a spectral width of 10330.6 Hz, an acquisition time of 3.17 s, and a recycle delay of 1.0 s per scan. Small amounts of AcOH/CDCl3 solution were added gradually to the BCD solutions (0.005 g in 0.7 mL of CDCl3). 1H NMR signals were assigned based on substitute effect, signal multiplicity, the magnitude of couplings constants and Nuclear Overhauser Effect experiments.
III. Results and discussion
Harmine
Fig. 1 shows the absorption spectra of HI in dioxane, in the presence of increasing quantities of AcOH. As can be seen, addition of low concentrations of a hydrogen bond donor [0–1% (v/v) of AcOH] (Fig. 1A) had a slight hyperchromic effect on the absorption maximum, with no appreciable shifts in its position. At higher donor concentrations, the absorption bands were slightly red-shifted (Fig. 1B).![Absorption spectra for HI in dioxane, with increasing quantities of AcOH. (A) 0–1% and (B) 1.2–9.2% (v/v). The insert shows the linear fit obtained by plotting log[A361/(Ac − A361nm)] vs. log(%AcOH).](/image/article/2011/RA/c1ra00205h/c1ra00205h-f1.gif) | ||
| Fig. 1 Absorption spectra for HI in dioxane, with increasing quantities of AcOH. (A) 0–1% and (B) 1.2–9.2% (v/v). The insert shows the linear fit obtained by plotting log[A361/(Ac − A361nm)] vs. log(%AcOH). | ||
Plotting  (where A361 represents the absorbance at different AcOH concentrations and Ac is the absorbance for the complex at a concentration equal to the initial concentration of HI) yielded a good linear fit, with a slope equal to 2: confirming a 1
(where A361 represents the absorbance at different AcOH concentrations and Ac is the absorbance for the complex at a concentration equal to the initial concentration of HI) yielded a good linear fit, with a slope equal to 2: confirming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio between HI and AcOH in the complex. Both absorbance values were measured at 361 nm, (see insert of Fig. 1B).
:2 ratio between HI and AcOH in the complex. Both absorbance values were measured at 361 nm, (see insert of Fig. 1B).
Related changes were also observed in the corresponding fluorescence spectra (see Fig. 2). Addition of small amounts of acetic acid caused a reduction in fluorescence intensity, coupled with a progressive red-shift. Interestingly, this red-shift ceases when the wavelength corresponding to the maximum of the cationic species (∼414 nm) is reached (i.e. when the pyridinic nitrogen is protonated). Between 0–0.1% (v/v) AcOH, the fluorescence intensity also displays a loss of vibrational structure. However, at AcOH concentrations greater than 0.5% (v/v), the decrease in intensity is greater, and a new fluorescence band at around 515 nm appears, which is hypsochromically shifted with increasing AcOH concentrations. At AcOH concentrations between 0–2% (v/v), analysis of fluorescence data for the HI/AcOH system in dioxane, revealed a linear dependence of 1/(I0 − I) on the reciprocal of AcOH concentration (where I0 is the initial fluorescence intensity of free HI at the 360 nm titration wavelength). According to the Benesi–Hildebrand equation, a 1:1 complex is formed at low AcOH concentrations. At higher AcOH concentrations, a new complex is formed, which is likely to be a 1:2 or even higher order complex.
 | ||
| Fig. 2 Fluorescence spectra of HI in dioxane, with increasing quantities of AcOH (%AcOH v/v). The insert shows the normalised spectra. | ||
Although similar behaviour was observed when chloroform was used, this solvent favours the observed prototropism, and requires lower concentrations of AcOH to obtain the same fluorescence profiles.
Thus, weak hydrogen bonding interactions clearly take place between AcOH and HI, which become stronger upon excitation. Moreover, this excitation-dependent increase in interaction strength is due to photo-induced electronic distribution changes, which greatly increase the pyridinic nitrogen basicity and pyrrolic nitrogen acidity in the S1 state.
In order to understand how the system evolves in the ground state upon addition of AcOH, 1H NMR spectra of HI were recorded in CDCl3 with increasing concentrations of AcOH (Fig. 3A). Fig. 3B shows plots of the chemical shift variation (Δδ) versus the molar ratio (ρ = [AcOH]/[BCD]): Δδ = δC − δF, where δF is the chemical shift in the absence of AcOH, and δC is the chemical shift after addition of AcOH. Consequently, Δδ for a specific proton is positive if addition of acetic acid causes deshielding at the electronic environment of the proton, while the opposite is true if Δδ is negative. As can be seen, we observed a larger Δδ value for the pyrrolic proton (Δδ ≈ 2 ppm) at an [AcOH]/[HI] molar ratio of 9.3. Thus, addition of AcOH decreases the electronic environment of this proton, resulting in a downfield chemical shift. Simultaneously, a small Δδ variation was observed for H3 (−0.30 ppm) and H4 (−0.25 ppm), both of which are located in the pyridinic ring. Peaks for both the H3 and H4 protons were shifted to higher fields (i.e. more shielded); possibly due to the formation of hydrogen bonds between the hydrogen atom of AcOH and the pyridinic nitrogen atom of HI. This interaction would shift the molecular electronic charge toward the pyridinic ring, increasing its electronic density.
![(A) 1H NMR spectra of HI in CDCl3, with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[HI]). (B) Chemical shift variation (Δδ) for HI versus molar ratio of AcOH/[HI] (Δδ = δC − δF, where δF is the chemical shift in the absence of AcOH and δC is the chemical shift after addition of AcOH).](/image/article/2011/RA/c1ra00205h/c1ra00205h-f3.gif) | ||
| Fig. 3 (A) 1H NMR spectra of HI in CDCl3, with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[HI]). (B) Chemical shift variation (Δδ) for HI versus molar ratio of AcOH/[HI] (Δδ = δC − δF, where δF is the chemical shift in the absence of AcOH and δC is the chemical shift after addition of AcOH). | ||
The chemical shifts of protons H6 and H8 (see Fig. 3B) are nearly unchanged, demonstrating that addition of AcOH does not significantly affect the electronic environment of these atoms. Similar behaviour was reported for harmane, another BCD.56 However, harmane exhibited larger shifts for H3 and H4 than HI, suggesting the formation of a cyclic 1:3 hydrogen-bond complex between harmane and AcOH, which does not occur between HI and AcOH.
2-Methylharmine
The spectroscopic properties of 2MHI were also similar in both dioxane and chloroform. However, the absorption spectra of 2MHI (model of T2 tautomer) were quite different from those observed for HI (model of T1 tautomer). As can be seen in Fig. 4, the spectrum is strongly red-shifted (with the band corresponding to the S1 state centred at 420 nm, and that for the S2 state at 360 nm) relative to that of HI, with a very low molar absorption coefficient, which can be associated with an nπ type singlet state. In addition, this absorption profile disappears upon addition of small amounts of acetic acid [<0.025% (v/v)], resulting in spectra that are similar to those observed for HI at high AcOH concentrations; and demonstrating the high capacity of the pyrrolic nitrogen of T2 to form hydrogen bonds.![Absorption spectra for 2MHI in dioxane, with increasing quantities of AcOH (%AcOH v/v). The insert shows the linear fit obtained by plotting log[(A0/A272nm) − 1] vs. log(%AcOH) at 272 nm.](/image/article/2011/RA/c1ra00205h/c1ra00205h-f4.gif) | ||
| Fig. 4 Absorption spectra for 2MHI in dioxane, with increasing quantities of AcOH (%AcOH v/v). The insert shows the linear fit obtained by plotting log[(A0/A272nm) − 1] vs. log(%AcOH) at 272 nm. | ||
Increasing AcOH concentrations increase the solvent polarity, resulting in blue-shifting of the absorption band and leading to the appearance of isosbestic points, which are indicative of an equilibrium. This equilibrium was analysed by considering that a new complex could be formed between the 2MHI/AcOH complex and another AcOH molecule. The validity of this possibility was investigated by plotting log−[(A0/A) − 1] vs. log (%AcOH) at 272 nm, where A0 is the absorbance at 272 nm and 0.025% (v/v) AcOH. This wavelength was selected because absorbance values were practically unchanged at large wavelengths. Using the above equation, we obtained a good linear fit, with a slope equal to 1, confirming the formation of a new complex (see insert of Fig. 4).
In fluorescence emission spectra, 2MHI displays a weak fluorescence band around 500 nm, the intensity of which increases with increasing AcOH in solution (dioxane or chloroform). This increase in fluorescence intensity of the band centred at 500 nm (F1), is accompanied by the appearance of a new blue-shifted fluorescence band, labelled F2, which has a maximum around 426 nm. As can be seen in Fig. 5, the spectral locations and shapes of these high-energy emissions (F2) coincide almost exactly with the fluorescence of cationic species recorded in pure AcOH. These findings confirm that the basicity of the five-membered ring nitrogen decreases during the transition from the ground to the S1 excited state. The hypsochromic shift of the F1 band observed when the polarity increases may be related to a strong nπ contribution to the electronic state of the molecule.
 | ||
| Fig. 5 Fluorescence spectra of 2MHI in dioxane, with increasing quantities of AcOH (%AcOH v/v). | ||
Fig. 6A shows 1H NMR spectra for 2MHI in the presence and absence of AcOH. Unlike HI, where the chemical shifts vary gradually with AcOH concentration, a more dramatic change was detected for 2MHI , indicating that even small amounts of AcOH strongly change the electronic distribution of this molecule. Table 1 compares the chemical shift values of all protons in 2MHI in the absence of AcOH, with all protons from a 1.7 molar ratio [AcOH]/[2MHI] mixture.
![(A) 1H NMR spectra of 2MHI in CDCl3, with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[2MHI]), (B) Δδ = δC − δ1.7versus molar ratio of [AcOH]/[2MHI] (δ1.7 is the proton chemical shift at [AcOH]/[2MHI] = 1.7).](/image/article/2011/RA/c1ra00205h/c1ra00205h-f6.gif) | ||
| Fig. 6 (A) 1H NMR spectra of 2MHI in CDCl3, with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[2MHI]), (B) Δδ = δC − δ1.7versus molar ratio of [AcOH]/[2MHI] (δ1.7 is the proton chemical shift at [AcOH]/[2MHI] = 1.7). | ||
| Proton | δ neutral species | δ complex | δ complex − δneutral |
|---|---|---|---|
| H3 | 7.46 | 7.77 | 0.31 |
| H4 | 7.88 | 7.90 | 0.02 |
| H5 | 7.98 | 7.92 | −0.06 |
| H6 | 6.81 | 6.94 | 0.13 |
| H8 | 7.33 | 7.56 | 0.23 |
| NCH3 | 4.17 | 4.19 | 0.02 |
| OCH3 | 3.94 | 3.98 | 0.04 |
| CH3 | 3.18 | 3.25 | 0.07 |
As can be seen from the fourth column of Table 1, the H3 proton appears to be more affected by the formation of the complex; indicating that the charge distribution of the molecule has been altered. Based on this evidence, we hypothesised that protonation of Npr induces a charge transfer from the pyridine ring to this nitrogen, causing deshielding of H3. To further investigate this behaviour, Δδ was plotted vs. the molar ratio of [AcOH]/[2MHI] (see Fig. 6B). Note that in this case, Δδ is Δδ = δC − δ1.7, where δF has been replaced by δ1.7 (i.e. the proton chemical shifts at AcOH/2MHI molar ratio of 1.7). As can be seen, the greatest variation again occurs in H3, but, unlike with HI, deshielding is now observed, because hydrogen bonding involving the five-membered nitrogen atom causes a charge transfer from the pyridinic to the pyrrolic nitrogen.
Harmaline
As can be seen in Fig. 7, addition of increasing quantities of AcOH to HLINE in dioxane solution results in the progressive appearance of a new band at 374 nm in the absorption spectra, with an isosbestic point that suggests the formation of a hydrogen-bonding complex. In chloroform, however, these progressive changes were not observed, since the addition of even very small amounts of AcOH leads to the sudden disappearance of the uncomplexed species and the appearance of the band at 374 nm. This fact demonstrates that loss of aromaticity in the pyridine ring greatly increases the basicity of its nitrogen.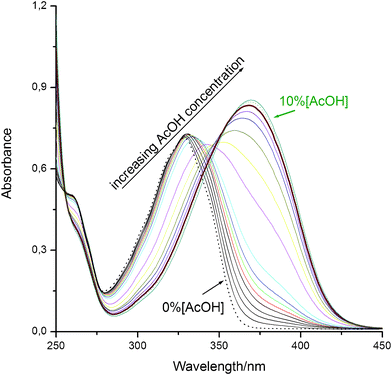 | ||
| Fig. 7 Absorption spectra for HLINE in dioxane with increasing quantities of AcOH (%AcOH v/v). | ||
Plotting  (where A381 represents absorbance values at 381 nm at different AcOH concentrations, and Ac is the absorbance at 381 nm of the complex at a concentration equal to the initial concentration of HLINE) resulted in a good linear fit, with a slope equal to 2, confirming a 1:2 relationship between HLINE and AcOH in the complex.
(where A381 represents absorbance values at 381 nm at different AcOH concentrations, and Ac is the absorbance at 381 nm of the complex at a concentration equal to the initial concentration of HLINE) resulted in a good linear fit, with a slope equal to 2, confirming a 1:2 relationship between HLINE and AcOH in the complex.
Fluorescence emission spectra were recorded for HLINE in both solvents. In the absence of AcOH, the Stokes' shift between absorption and emission spectra was approximately 130 nm; suggesting that the emitting species is not the same as the absorbing species. Since the basic properties of the pyridine nitrogen in these compounds increases upon electronic excitation to S1, we associated this shift to the formation of a complex with the solvent.
Addition of small amounts of AcOH produces an increase in fluorescence intensity and a progressive red-shift (approximately 20 nm in chloroform), followed by a blue-shift that continues until about 460 nm, the wavelength corresponding to the maximum of the cationic species (see Fig. 8).
 | ||
| Fig. 8 Fluorescence spectra of HLINE in dioxane with increasing quantities of AcOH (%AcOH v/v). | ||
1H NMR spectra for HLINE are shown in Fig. 9A. Based on a plot of Δδ versus the molar ratio of [AcOH]/[HLINE] (see Fig. 9B), the largest chemical shift changes occur for the hydrogen of the Npr–H group after addition of small amounts of AcOH (Δδ = 4 ppm). These results indicate that this nitrogen is much more acidic in HLINE than in other BCDs, facilitating stronger interactions with AcOH.
![(A) 1H NMR spectra of HLINE in CDCl3 with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[HLINE]). (B) Δδ versus molar ratio of [AcOH]/[HLINE] (Δδ = δC − δF, where δF is the chemical shift in the absence of AcOH and δC is the chemical shift after addition of AcOH).](/image/article/2011/RA/c1ra00205h/c1ra00205h-f9.gif) | ||
| Fig. 9 (A) 1H NMR spectra of HLINE in CDCl3 with increasing quantities of AcOH (ρ is the molar ratio of [AcOH]/[HLINE]). (B) Δδ versus molar ratio of [AcOH]/[HLINE] (Δδ = δC − δF, where δF is the chemical shift in the absence of AcOH and δC is the chemical shift after addition of AcOH). | ||
As can be seen, the electronic environments of the methylene protons (H3) and methyl hydrogens in HLINE change in opposite direction: H3 becomes shifted to lower frequencies, while the methyl protons are shifted to higher values. The chemical shift variations observed for the methylene group at position 3, upon addition of AcOH to HLINE in chloroform, may be due to the formation of a hydrogen bond between AcOH and the pyridinic nitrogen atom. Although this behaviour is similar to that observed for HI, the interaction between AcOH and the pyrrolic nitrogen is stronger in HLINE than in HI.
In agreement with our UV-Vis results, that suggest a 1:2 complex, the NMR signals of pyrrol are strongly shifted to higher frequencies. Based on these results, we propose a double interaction between two molecules of AcOH and one molecule of HLINE, involving the two nitrogen atoms. As in HI, the Npd⋯HOAc interaction shifts the molecular electronic charge density toward the pyridine ring. At the same time, the strong Npr–H⋯O interaction increases the negative charge density on the pyrrolic nitrogen, favouring charge transfer to the pyridine ring in the S1 state. In other words, there is a synergetic effect from two forces which increases HLINE basicity in both the ground and the first excited state.
The above results prove the existence of interactions between the BCD compounds analysed and AcOH. In HI and HLINE, these interactions occur through the two nitrogen atoms, while in 2MHI, they are only possible through the pyrrolic ring.
The lone electron pair on Npd interacts with the hydroxyl group of AcOH, producing a positive charge density on this nitrogen, which is compensated by attracting the π-cloud; consistent with the observed shielding of H3 in HI and HLINE. In agreement with this, the opposite effect was observed in 2MHI because the Npd⋯HO interaction is not possible. In this case, the hydroxyl group of AcOH interacts with the lone electron pair on the pyrrolic nitrogen atom, giving rise to a positive charge density on this nitrogen. This interaction involves displacement of the π-cloud toward the five-membered ring, and consequently deshielding of H3 and an increase in methyl group charge density.
Furthermore, the interaction between the Npr–H group and the lone electron pair on the AcOH carbonyl group leads to a negative charge density in both HI and HLINE, but not in 2MHI. This increased π-cloud electron density contributes to the induced charge transfer from the pyrrolic to the pyridinic ring, and consequently, the increased basicity of Npd. Accordingly, the greater the Npr–H⋯O interaction the greater the negative charge density, and therefore the greater the basicity of Npd.
Our NMR results confirm this hypothesis. For example, in HLINE, the observed shift for Npr–H due to the addition of AcOH was higher than that observed for HI. These findings are consistent with the presence of a stronger Npr–H⋯O interaction, which causes HLINE to be more acidic than HI.
We propose that the key to understanding this system is to consider an equilibrium between two tautomers, T1 (represented by HI) and T2 (represented by 2MHI). The excess electronic density in the Npd atom increases its basicity, favouring the interaction between its lone electron pair and the hydrogen donor molecule. With increasing concentrations of AcOH in solution, the Npd⋯H distance decreases until it becomes short enough to form the Npd–H bond. At this point, the Npd lone electron pair is coupled to the π-cloud, resulting in a quinoid type structure, which is associated with the T2 tautomer. The subsequent hypsochromic shift of T2 fluorescence, observed when solvent polarity increases, confirms the strong participation of the nπ state in this system.
T1 ↔ T2 equilibrium only occurs in the excited state, in low polar media and in the absence of protons. The relative positions of the T1 and T2 minima in the S1 state vary with the polarity of the medium. Furthermore, the T2 minimum disappears in polar media, indicating strong involvement of the π-cloud in the photo-tautomerisation process, and confirming the hypothesis established in previous papers that polar media inhibit the photo-tautomerisation process.
IV. Conclusions
In order to explain the mechanism of Z/Q formation (and the emission band at 520 nm), we propose an oscillating system between T1 and T2 tautomers, whose equilibrium is influenced by several processes. Specifically, upon excitation, a redistribution of the π electron cloud occurs via intramolecular charge transfer from the pyrrolic to the pyridinic ring. This charge transfer reduces the electronic density in the Npr, weakening the Npr–H bond and facilitating proton dissociation. In contrast, excess electronic density in Npd increases its basicity, favouring the interaction between its lone electron pair and the hydrogen donor molecule. Furthermore, when the Npd-donor bond is sufficiently short, the lone electron pair orbital of Npd is coupled to the π-cloud by an out-of-plane vibration, resulting in a quinoid type structure associated with the T2 tautomer. At this point, charge transfer begins to occur from the pyridinic to the pyrrolic ring. Once the charge is transferred to Npr, this nitrogen becomes more basic and Npd becomes less basic. In this case, coupling occurs between the lone electron pair orbital of Npr and the π-cloud, resulting again T1. Therefore, the electronic transfer can take place in both directions: from the pyrrolic to pyridinic ring for the zwitterionic structure isomer, or from the pyridinic to pyrrolic ring for the quinoid structure. This balance between T1 and T2 only occurs in low polarity media, and in the absence of protons; while the presence of protons stabilises the system and prevents charge transfer.Acknowledgements
We thank Fundacion Carolina the fellowship of F. Hallwass.References
- P. T. Chou, J. Chinese Chem. Soc., 2001, 48, 651 CAS.
- C. A. Taylor, A. El-Bayoumi and M. Kasha, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 253 CrossRef CAS.
- P. Avouris, L. L. Yang and M. A. El-Bayoumi, Photochem. Photobiol., 1976, 24, 211 CrossRef CAS.
- A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 378, 260 CrossRef CAS.
- M. Chaschisvilis, T. Fiebig, A. Douhal and A. H. Zewail, J. Phys. Chem. A, 1998, 102, 669 CrossRef.
- S. Takeuchi and T. Tahara, J. Phys. Chem. A, 1998, 102, 7740 CrossRef CAS.
- R. Lopez-Martens, P. Long, D. Solgadi, B. Soep, J. Syage and Ph. Millie, Chem. Phys. Lett., 1997, 273, 219 CrossRef CAS.
- A. Nakajima, M. Hirano, R. Hasumi, K. Kaya, H. Watanabe, C. C. Carter, J. M. Williamson and T. A. Miller, J. Phys. Chem. A, 1997, 101, 392 CrossRef CAS.
- A. V. Smirnov, D. S. English, R. L. Rich, J. Lane, L. Teyton, A. W. Schwabacher, S. Luo, R. W. Thornburg and J. W. Petrich, J. Phys. Chem. B, 1997, 101, 2758 CrossRef CAS.
- D. McMorrow and T. Aartsma, Chem. Phys. Lett., 1986, 125, 581 CrossRef CAS.
- J. Konijnenberg, A. H. Huizer and C. A. G. O. Varma, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1163 RSC.
- R. S. Moog and M. Maroncelli, J. Phys. Chem., 1991, 95, 10359 CrossRef CAS.
- J. Herbich, J. Sepiol and J. Waluk, J. Mol. Struct., 1984, 114, 329 CrossRef CAS.
- C. F. Chapman and M. Maroncelli, J. Phys. Chem., 1992, 96, 8430 CrossRef CAS.
- S. Mente and M. Maroncelli, J. Phys. Chem. A, 1998, 102, 3860 CrossRef CAS.
- Y. Chen, F. Gai and J. W. Petrich, J. Am. Chem. Soc., 1993, 115, 10158 CrossRef CAS.
- Y. Chen, F. Gai and J. W. Petrich, Chem. Phys. Lett., 1994, 222, 329 CrossRef CAS.
- P.-T. Chou, M. L. Martinez, W. C. Cooper, S. T. Collins, D. P. McMorrow and M. Kasha, J. Phys. Chem., 1992, 96, 5203 CrossRef CAS.
- Y. Chen, R. L. Roich, F. Gai and J. W. Petrich, J. Phys. Chem., 1993, 97, 1770 CrossRef CAS.
- C.-P. Chang; H. Wen-Chi, K. Meng-Shin, P.-T. Chou and J. H. Clemens, J. Phys. Chem., 1994, 98, 8801 CrossRef.
- P.-T. Chou, C.-Y. Wei, C.-P. Chang and K. Meng-Shin, J. Phys. Chem., 1995, 99, 11994 CrossRef CAS.
- T. Fiebig, M. Chaschisvilis, M. Manger, A. H. Zewail, A. Douhal, I. Garcia-Ochoa and A. D. H. Ayuso, J. Phys. Chem. A, 1999, 103, 7419 CrossRef CAS.
- M. Kasha, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2379 RSC.
- M. Kasha, Acta Phys. Pol. A, 1987, 71, 717 Search PubMed.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1 CrossRef CAS.
- S. J. Formosinho and L. G. Arnaut Photochem., J. Photochem. Photobiol., A, 1993, 75, 21 CrossRef CAS.
- S. M. Ormson and R. G. Brown, Prog. React. Kinet., 1994, 19, 45 CAS.
- D. Le Gourrierec, S. M. Ormson and R. G. Brown, Prog. React. Kinet., 1994, 19, 211 CAS.
- M. M. Airaksinen and I. Kari, Med. Biol., 1981, 59, 21 CAS.
- T. S. Zhou, W. C. Ye and Z. T. Wang, Phytochemistry, 1998, 49, 1807 CrossRef CAS.
- G. M. Cabrera and A. M. Seldes, J. Nat. Prod., 1999, 62, 759 CrossRef CAS.
- S. Kotanen, J. Huybrechts and A. Cerstiaens, Biochem. Biophys. Res. Commun., 2003, 310, 64 CrossRef CAS.
- S. Fukushima, K. Matsubara, A. Akane and H. Shiono, Alcohol, 1992, 9, 31 CrossRef CAS.
- S. Manabe, J. Yuan, T. Takahashi and R. C. Urban Jr, Exp. Eye Res., 1996, 63, 179 CrossRef CAS.
- O. Beck and A. Lundman, Biochem. Pharmacol., 1983, 32, 1507 CrossRef CAS.
- J. Adachi, Y. Mizoi, T. Naito, Y. Ogawa, Y. Uetani and I. Ninomiya, J. Nutr., 1991, 121, 646 CAS.
- K. Hayashi, M. Nagao and T. Sugimura, Nucleic Acids Res., 1977, 4, 3679 CrossRef CAS.
- D. Csanyi, G. Hajos and Z. Riedl, Bioorg. Med. Chem. Lett., 2000, 10, 1767 CrossRef CAS.
- Y. Song, J. Wang, S. F. Teng, D. Kesuma, Y. Deng, J. Duan, J. H. Wang, R. Zhong-Qi and M. M. Sim, Bioorg. Med. Chem. Lett., 2002, 12, 1129 CrossRef CAS.
- H. Nii, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2003, 541, 123 CrossRef CAS.
- A. M. Deveau, M. A. Labroli and C. M. Dieckhaus, Bioorg. Med. Chem. Lett., 2001, 11, 1251 CrossRef CAS.
- H. Kim, O. S. Sablin and R. R. Ramsay, Arch. Biochem. Biophys., 1997, 337, 137 CrossRef CAS.
- N. S. Buckholtz and W. O. Boggan, Biochem. Pharmacol., 1977, 26, 1991 CrossRef CAS.
- C. Braestrup, M. Nielsen and C. E. Olsen, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 2288 CrossRef CAS.
- W. Schlecker, A. Huth, E. Ottow and J. Mulzer, Synthesis-Stuttgart, 1995,(10), 1225 CrossRef CAS.
- A. Batch and R. H. Dodd, J. Org. Chem., 1998, 63, 872 CrossRef CAS.
- P. Molina, P. M. Fresneda and S. Garcia-Zafra, Tetrahedron Lett., 1995, 36, 3581 CrossRef CAS.
- P. Molina, P. M. Fresneda, S. Garcia-Zafra and P. Almendros, Tetrahedron Lett., 1994, 35, 8851 CrossRef CAS.
- D. Reyman, A. Pardo and J.-M.-L. Poyato, J. Phys. Chem. A, 1994, 98, 10408 CAS.
- D. Reyman and M. H. Viñas, Chem. Phys. Lett., 1999, 301, 551 CrossRef CAS.
- A. Dias, A. P. Varela, M. D. Miguel, A. L. Macanita and R. S. Becker, J. Phys. Chem., 1992, 96, 10290 CrossRef CAS.
- C. Carmona, M. Balon, A. S. Coronilla and M. A. Munoz, J. Phys. Chem. A, 2004, 108(11), 1910 CrossRef CAS.
- A. Sanchez-Coronilla, M. Balón, M.-A. Muñoz and C. Carmona, Chem. Phys., 2008, 344, 72 CrossRef CAS.
- A. Sanchez-Coronilla, M. Balón, M.-A. Muñoz, J. Hidalgo and C. Carmona, Chem. Phys., 2008, 351, 27 CrossRef CAS.
- J. Hidalgo, A. Sanchez-Coronilla, M. Balón, M.-A. Muñoz and C. Carmona, Photochem. Photobiol. Sci., 2009, 8, 414 CAS.
- D. Reyman, F. Hallwass, S. M. Gonçalves and J.-J. Camacho, Magn. Reson. Chem., 2007, 45, 830 CrossRef CAS.
- P.-T. Chou, Y. I. Liu and G. R. Wu, J. Phys. Chem. B, 2001, 105, 10674 CrossRef CAS.
- A. Sanchez-Coronilla, C. Carmona, M.-A. Muñoz, J. Hidalgo and M. Balón, Chem. Phys., 2006, 327, 70 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |