Understanding corrosion inhibition mechanisms—experimental and theoretical approach
Emeka E.
Oguzie
*ab,
Ying
Li
*a,
Sheng G.
Wang
c and
Fuhui
Wang
a
aState Key Laboratory for Corrosion and Protection, Institute of Metal Research, Chinese Academy of Sciences, 62 Wencui Road, Shenyang, 110016, China. E-mail: emekaoguzie@gmail.com; liying@imr.ac.cn; Tel: +234 803 7026581 Tel: +86 24 23992875
bElectrochemistry and Materials Science Research Laboratory, Department of Chemistry, Federal University of Technology Owerri, PMB 1526, Owerri, Nigeria
cShenyang National Laboratory for Materials Sciences, Institute of Metal Research and International Centre for Materials Physics, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang, 110016, China
First published on 13th September 2011
Abstract
We describe a combined electrochemical and first principle density functional study to probe the corrosion inhibiting and adsorption behavior of methionine (Met) and phenylalanine (Phe) on polycrystalline and nanocrystalline iron in acid media. Met functioned as a better inhibitor for both Fe microstructures, and was more favorably adsorbed on the nanocrystalline surface. The nanocrystalline surface however diminished adsorption of Phe. The comparable values of our computed physisorption energies (−94.2 kcal mol−1 and −86.6 kcal mol−1 for Phe and Met respectively) as well as the stable adsorption orientations of both molecules on Fe suggest a controlling influence of a soft epitaxial adsorption mechanism in which C, N, O, S atoms of the molecules align with epitaxial grooves on the Fe lattice. The significant contribution of physisorptive interactions also correlates with the similarity in experimental inhibition efficiencies on polycrystalline Fe (Phe = 73% and Met = 82%), though for Met the thiol group imparts an added ability for covalent interaction with Fe, which accounts for the higher efficiency. Furthermore, we have related the diminished inhibition efficiency of Phe on the nanocrystalline Fe surface to disruption of the epitaxial patterns on the lattice as the surface becomes increasingly defective, leading to weaker adsorption. The improved efficiency of Met on the nanocrystalline surface is related to scaling up of the covalent interactions around defect sites. Our theoretical conclusions are validated by the consistency with our experimental findings.
1. Introduction
The corrosion phenomenon is globally recognized as a significant challenge of modern civilization and consequently the development of corrosion control measures and techniques remains a topic of important industrial and academic concern. The basic objective of corrosion inhibitor research is to gain insight into the mechanisms by which materials added to a fluid aggressive environment retard the metal–corrodent reaction. In spite of the widespread and sustained interest in the investigation and application of organic (adsorption-type) inhibitors, the results obtained so far reveal that the inhibition process is neither uniform with respect to all the classes of compounds studied nor even constant or consistent with one inhibitor in a given system. Indeed, the effectiveness of the overall process is a function of the metal, corrodent, molecular and electronic structure and concentration of the inhibitor, as well as temperature and other environmental considerations.1–10 This seeming complexity is the reason why there is as yet no universally acceptable consensus regarding the actual mechanism of the corrosion inhibition process and provides the motivation for more extensive studies involving combinations of conventional and novel ideas and methodologies.A practical route to study the complex processes associated with metal–inhibitor interactions at the molecular level involves computer simulations of suitable models and the density functional theory (DFT) has been used widely in this regard. Recent advances in DFT-based quantum chemical computations have made this powerful tool increasingly available to corrosion scientists for theoretical investigation of corrosion and corrosion inhibition systems. Such computations have been widely used to analyze the molecular electronic structures of a wide range of organic inhibitors using a number of quantum chemical descriptors.10–20 Recently, attention has focused on modeling the interaction of inhibitor molecules with metal surfaces and computing the interaction energies, taking cognizance of both covalent and non-covalent contributions to the interaction energy.20–24 Such an approach offers the added advantage of providing important physical insights on corrosion inhibition mechanisms.
The ability to process and investigate materials at the nano-scale makes for better understanding of fundamental physicochemical properties, which can provide vital insights into the mechanisms of metal corrosion and protection, particularly since variations in particle size and metal microstructure determine the rate of diffusion of both the corrodent and inhibitor species through the lattice. A number of studies have examined the corrosion behavior of nanocrystalline materials with contradictory results which have enriched our understanding of the mechanism of corrosion reactions in different environments.25 Much of this is captured in a recent review article on the corrosion behavior of nanocrystalline materials by Li and co-workers.26 On the other hand, only little information is available on the corrosion inhibition of nanocrystalline metals and alloys,27,28 which means that any new mechanistic information derivable from such interactions is yet to be fully realized.
Coupling key ideas in nanotechnology with recent advances in DFT-based quantum chemical computations will enable better understanding of key phenomena associated with the corrosion inhibition process. This reasoning is in line with the current direction in contemporary physical chemistry to converge experimental and theoretical approaches for analysis of sufficiently complex model systems.29 Our current attempts at elucidating the mechanisms of iron–inhibitor (Fe–Inh) interactions involves assessment of the effect of metal microstructure modification on corrosion inhibitor adsorption and hence performance. In this paper we employ experimental and DFT techniques to investigate the inhibiting effects of phenylalanine (Phe) and methionine (Met) on the corrosion of polycrystalline iron (grain size ∼50 μm) and nanocrystalline iron (∼39 nm) specimens in sulfuric acid. The motivation for the computational studies is not so much to explain specific data for each system, but rather to give a theoretical framework in which to understand the relative magnitudes and qualitative behavior of the interactions and how these are modified by altering the metal surface microstructure. We have chosen two amino acids with distinctive differences in molecular structure (the phenyl ring in Phe and thiol group in Met) in order to understudy how the different functional groups interact with polycrystalline and nanocrystalline Fe.
2. Computational details
All theoretical computations were performed within the framework of DFT using the Materials Studio; MS Modeling 4.0 software (Accelrys Inc.). The electronic structures of Met, Phe and the Fe surface were modeled by means of the DFT electronic structure program DMol3 using a Mulliken population analysis as well as a Hirshfeld numerical integration procedure.30,31 Electronic parameters for the simulation include restricted spin polarization using the DNP basis set and the Perdew Wang (PW) local correlation density functional. The distribution of frontier molecular orbitals and Fukui indices were assessed for the inhibitor molecules, with a view to establishing the active sites as well as local reactivity of the molecules.Molecular dynamics (MD) simulation of the non-covalent interaction between a single inhibitor molecules and the Fe surface was performed using Forcite quench molecular dynamics to sample many different low energy configurations and identify the low energy minima.32,33 Calculations were carried out, using the COMPASS force field and the Smart algorithm, in a simulation box 30 Å × 25 Å × 29 Å with periodic boundary conditions to model a representative part of the interface, devoid of arbitrary boundary effects. The box was comprised of the Fe slab, cleaved along the (110) plane and a vacuum layer of 20 Å height. The geometry of the bottom layer of the slab was constrained to the bulk positions whereas other degrees of freedom were relaxed before optimizing the Fe (110) surface, which was subsequently enlarged into a 10 × 8 supercell. Inhibitor molecules were adsorbed on one side of the slab. Temperature was fixed at 303 K, with NVE (microcanonical) ensemble, with a time step of 1 fs and simulation time 5 ps. The system was quenched every 250 steps. Optimized structures of Phe, Met and the Fe surface were used for the simulation.
Covalent interactions between Met and the Fe (110) surface was carried out in a simulation box (7.45 Å × 7.45 Å × 22.03 Å) with periodic boundary conditions. The Fe (110) was first built and relaxed by minimizing its energy via molecular mechanics using the Discover molecular simulation program (MS Studio 4.0). The surface area was increased and its periodicity was changed by constructing a 5 × 4 super cell, with a vacuum slab of thickness 20 Å. We used the DMol3 code to study the chemisorption of Met onto the Fe (110) surface. Structure optimizations and corresponding total energy calculations of the most stable geometries are based on the generalized-gradient approximation (GGA) function with the Perdew–Burke–Ernzerhof (PBE) correction.34 For core electrons in the lowest lying atomic orbitals, the DFT semicore pseudopotentials (DSPP) core treatment, which replaces core electrons by a single effective potential, was implemented for relativistic effects.35 The Dmol3 electronic options were adjusted as follows; Monkhorst–Pack k-point mesh parameters were set to 2 × 2 × 1, with k point separation 0.05 Å−1. Self consistent field procedures were carried out with a convergence criterion of 10−5, using direct inversion in an iterative subspace (DIIS) and orbital occupancy smearing parameter of 0.005 Ha to speed up SCF convergence. We have neglected solvent and charge effects in all our simulations and performed the calculations at the metal/vacuum interface. Although this is clearly an oversimplification of the factual situation, it is adequate to qualitatively illustrate the differences in the adsorption behaviour of both molecules and provide sufficient insight to rationalize our experimental findings.
3. Results and discussion
3.1. Experimental observations
The first consideration was to experimentally ascertain the corrosion inhibiting efficacy of Phe and Met on both Fe microstructures. This was achieved by means of polarization and impedance measurements. Fig. 1 illustrates typical potentiodynamic polarization curves (Fig. 1a and 1b) and impedance plots (Fig. 1c and 1d) of polycrystalline and nanocrystalline Fe in 0.5 M H2SO4 solution without and with Phe. Similar plots for Met are given in Fig. 2. The corresponding electrochemical parameters, including the corrosion potential (Ecorr), corrosion current density (icorr), polarization resistance (Rp) and the cathodic and anodic Tafel slopes (βa and βc) are given in Table 1.![Polarization (a, b) and impedance (c, d) plots of polycrystalline Fe (CPII: a, c) and nanocrystalline Fe (BNII: b, d) in 0.5 M H2SO4 without and with Phe [CPII = conventional polycrystalline ingot iron; BNII = bulk nanocrystalline ingot iron].](/image/article/2011/RA/c1ra00148e/c1ra00148e-f1.gif) | ||
| Fig. 1 Polarization (a, b) and impedance (c, d) plots of polycrystalline Fe (CPII: a, c) and nanocrystalline Fe (BNII: b, d) in 0.5 M H2SO4 without and with Phe [CPII = conventional polycrystalline ingot iron; BNII = bulk nanocrystalline ingot iron]. | ||
![Polarization (A, B) and impedance (C, D) plots of polycrystalline Fe (CPII: A, C) and nanocrystalline Fe (BNII: B, D) in 0.5 M H2SO4 without and with Met [CPII = conventional polycrystalline ingot iron; BNII = bulk nanocrystalline ingot iron].](/image/article/2011/RA/c1ra00148e/c1ra00148e-f2.gif) | ||
| Fig. 2 Polarization (A, B) and impedance (C, D) plots of polycrystalline Fe (CPII: A, C) and nanocrystalline Fe (BNII: B, D) in 0.5 M H2SO4 without and with Met [CPII = conventional polycrystalline ingot iron; BNII = bulk nanocrystalline ingot iron]. | ||
| System | E corr (mV, SCE) | i corr (μA cm−2) | β a (mV/dec) | β c (mV/dec) | R p (Ohm) | R ct (Ohm cm2) | Q (Ohm−1 sn cm−2 ×10−4) | n |
|---|---|---|---|---|---|---|---|---|
| Inhibition efficiency (η%) values are given in parentheses. | ||||||||
| PC-Fe | ||||||||
| Blank | −523 | 243 | 129 | 122 | 35.82 | 19.0 | 2.14 | 0.92 |
| Met | −517 | 45.6 (81) | 96 | 146 | 197.2 (82) | 114.0 (83) | 1.17 | 0.93 |
| Phe | −518 | 67.7 (72) | 99 | 148 | 163.9 (78) | 80.3 (76) | 1.74 | 0.93 |
| NC-Fe | ||||||||
| Blank | −534 | 705 | 202 | 153 | 17.9 | 15.1 | 1.98 | 0.92 |
| Met | −513 | 71.5 (90) | 77 | 136 | 169.8 (90) | 82.9 (82) | 1.34 | 0.94 |
| Phe | −534 | 288 (59) | 128 | 144 | 50.6 (64) | 38.2 (61) | 1.53 | 0.93 |
Potentiodynamic polarization experiments were undertaken to distinguish the inhibiting effect of the additives on the anodic and cathodic corrosion reactions of Fe and to ascertain how this may be influenced by the Fe microstructure. The similarity of the polarization curves for both specimens suggests comparable corrosion and corrosion inhibition mechanisms. The results show that nanocrystalline Fe was more susceptible to corrosion in the acidic environment, with higher corrosion current density. This behavior has been adequately accounted for in references 26 and 29. Both Phe and Met functioned as mixed-type inhibitors, affecting both the anodic and cathodic reactions of the corrosion process. Met shifted Ecorr of both CPII and BNII towards more positive (noble) values. Phe exerted similar effect on CPII but did not modify Ecorr of BNII.
Impedance experiments were undertaken to explore the characteristics and kinetics of electrochemical processes occurring at the metal/electrolyte interface in the absence and presence of additives. The impedance responses of polycrystalline and nanocrystalline Fe without and with the test inhibitors, measured at Ecorr after 30 min immersion are presented as Nyquist plots in Fig. 1 (c, d) and Fig. 2 (C, D) respectively for Phe and Met. The Nyquist plots show single semicircles for all systems over the frequency range studied, again implying that the nanostructured specimen behaves electrochemically as the conventional Fe. The plots also show that nano-processing caused a decrease in the size of the semicircle and the impedance of the interface implying reduced corrosion resistance, while introduction of the inhibitors had the reverse outcome, indicating an inhibiting effect.
The high frequency intercept with the real axis in the Nyquist plots is assigned to the solution resistance (Rs) and the low frequency intercept with the real axis ascribed to the charge transfer resistance (Rct). To obtain the numerical values of the various impedance parameters presented in Table 1, the impedance spectra were analyzed by fitting to the equivalent circuit model Rs(QdlRct), in which the solution resistance is shorted by a constant phase element (CPE) that is placed in parallel to the charge transfer resistance. The CPE is used in place of a capacitor to compensate for deviations from ideal dielectric behavior arising from the inhomogeneous nature of the electrode surfaces. The impedance of the CPE is given by ZCPE = Q−1(jω)−n, where Q and n represent the magnitude and exponent of the CPE respectively, j is an imaginary number and ω is the angular frequency in rad s−1. The data in Table 1 clearly show that both Met and Phe enhanced the Rct, confirming their corrosion inhibiting effect. The proportionality factor Qdl of CPE was also modified by inhibitor adsorption on the metal/electrolyte interface, yielding lower values than was observed in the absence of the inhibitors.
Lower Qdl values corresponds to reduced interfacial capacitance, which, according to the Helmholtz model (Cdl = εεoA/δ); results from a decrease in the dielectric constant (ε) or an increase in the double layer thickness (δ). ε is the dielectric constant of the medium, εo the vacuum permittivity, A the electrode area and δ the thickness of the interfacial layer. Since adsorption of an organic inhibitor on a metal surface involves the replacement of adsorbed water molecules on the surface (Inhsol + xH2Oads → Inhads + xH2Osol), the smaller dielectric constant of the organic molecule compared to water as well as the increased thickness of the double layer due to inhibitor adsorption act simultaneously to reduce the interfacial capacitance. This provides experimental evidence of adsorption of Phe and Met on polycrystalline and nanocrystalline Fe.
Efficiency of inhibition (η%) was quantified from icorr, Rp and Rct respectively by comparing values obtained in the absence and presence of the inhibitors:
 | (1) |
3.2. Theoretical considerations
In order to explore the molecular interactions in an exhaustive and rigorous manner, we have adopted two complementary approaches which involve assessment of the electron distribution in the interacting species in this study (Phe, Met, Fe) as well as modeling the adsorption structures of Phe and Met on the Fe surface. Certain electronic structure parameters have been correlated with the effectiveness of adsorption-type inhibitors. These include the energy of the highest occupied molecular orbital (HOMO), which is associated with the capacity of a molecule to donate electrons, the lowest unoccupied molecular orbital (LUMO) energy corresponding to a tendency for electron acceptance and the HOMO–LUMO energy gap. Others include charge densities, electronic energies, dipole moments, molecular surface area etc.15–24 The electronic structures of the interacting species in this study (i.e. Phe, Met and Fe) are shown in Fig. 3–5. The geometry optimized structures, Fukui function for electrophilic attack (f−), total electron density, HOMO and LUMO orbitals are presented in Fig. 3a–3e respectively, for Phe, and in Fig. 4a–4e for Met. Some of the calculated quantum chemical properties are given in Table 2. Fig. 5a and 5b show the total electron density and the HOMO orbital of the Fe (110) lattice and highlight a deficiency of electrons around the corner/edge Fe atoms.![Electronic properties of Phe: (a) optimized structure [C, gray; H, white; N, green; O, red]; (b) f−; (c) total electron density; (d) HOMO orbital; (e) LUMO orbital.](/image/article/2011/RA/c1ra00148e/c1ra00148e-f3.gif) | ||
| Fig. 3 Electronic properties of Phe: (a) optimized structure [C, gray; H, white; N, green; O, red]; (b) f−; (c) total electron density; (d) HOMO orbital; (e) LUMO orbital. | ||
![Electronic properties of Met: (a) optimized structure [C, gray; H, white; N, green; O, red; S, yellow]; (b) f−; (c) total electron density; (d) HOMO orbital; (e) LUMO orbital.](/image/article/2011/RA/c1ra00148e/c1ra00148e-f4.gif) | ||
| Fig. 4 Electronic properties of Met: (a) optimized structure [C, gray; H, white; N, green; O, red; S, yellow]; (b) f−; (c) total electron density; (d) HOMO orbital; (e) LUMO orbital. | ||
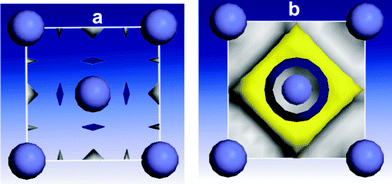 | ||
| Fig. 5 Electronic properties of the Fe (110) lattice: (a) total electron density, (b) HOMO orbital. | ||
| Property | Met | Phe |
|---|---|---|
| E HOMO (eV) | −5.52 | −5.48 |
| E LUMO (eV) | −1.76 | −1.43 |
| E LUMO–HOMO | 3.76 | 4.05 |
| Max. f− (Mulliken) | 0.503 (S) | 0.116 (N) |
| Max. f+ (Mulliken) | 0.203 (O) | 0.206 (C) |
| Total energy (eV) | −1876.98 | −2144.01 |
| Molecular weight (g mol−1) | 149.21 | 165.19 |
| Molecular surface area (Å2) | 189.9 | 216.28 |
The HOMO orbital of Phe is saturated around the amino function and the LUMO orbital is localized around the phenyl ring and carboxylate function, which can interact with the d-orbital of Fe using antibonding orbitals to form feedback bonds. For Met, the HOMO orbital is made up predominantly of the thiol group, while the LUMO density is largely around the carboxylate and amino groups. The local reactivity of each molecule was analyzed by means of the Fukui indices (FI) to assess reactive regions in terms of nucleophilic and electrophilic behavior. The f− measures reactivity with respect to electrophilic attack or the propensity of the molecule to release electrons, while f+ is a measure of reactivity relating to nucleophilic attack or tendency of the molecule attract electrons. The highest f− values (Mulliken analyses), associated with the S atom for Met and the N atom for Phe, indicate the sites most prone to nucleophilic attack and through which the molecules will interact with the metal. The high f+ values are associated with the hydroxyl O atom and the carbonyl C atom for Met and Phe respectively (both around the carboxylate function).
The data in Table 2 show that the two molecules display somewhat similar electronic structure parameters, with comparable values of the quantum chemical descriptors, the only notable difference being the high value of the f− function for Met, which means that the molecule will more readily form molecule–surface covalent bonds via the S atom, a common feature of many thio-containing amino acids and indeed organic molecules.36,37
We performed molecular dynamics (MD) simulations to understudy the interactions of single inhibitor molecules and the Fe surface. Metal–inhibitor interactions are categorized as either chemisorption (involving formation of molecule–surface covalent bonds) or physisorption (involving van der Waals dispersion forces). Chemisorbed molecules are thought to provide more effective protection since they reduce the inherent reactivity of the metal at the sites where they are attached. Physisorbed species function by creating a physical barrier to ingress of corrosive species and this becomes increasingly important for large molecules.
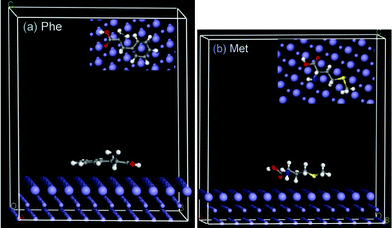 | ||
| Fig. 6 Representative snapshots of (a) Phe and (b) Met adsorbed on Fe (110). Inset images show the on-top views, emphasizing the soft epitaxial adsorption mechanism with accommodation of the molecular backbone in characteristic epitaxial grooves on the metal surface. | ||
To quantitatively appraise the interaction between each molecule and the Fe surface, the adsorption energy (Eads) was calculated using the relationship in eqn (2), wherein a negative value of Eads corresponds to a stable adsorption structure.
| Eads = Etotal − (EMol + EFe) | (2) |
Close inspection of the on-top view of adsorbed Phe and Met on Fe (110) reveals a very clear trend in the adsorption configuration of both molecules wherein polarizable atoms (C, N, O, S) along the molecular backbone align with vacant sites on the fcc lattice atop the metal surface and actually avoid contact with the Fe atoms on the surface plane (larger spheres on the Fe slab). This corresponds to accommodation of the molecular backbone in characteristic epitaxial grooves on the metal surface—the so-called soft epitaxial adsorption mechanism of Heinz and co-workers, which appropriately describe non covalent adsorption of amino acids on metal surfaces.36,38 Epitaxial adsorption orientations are associated with a minimum free energy of adsorption as the adsorbed molecules can be considered as extensions of the fcc lattice and adsorption strength scales with improved fit of the polarizable atoms of a molecule to multiple epitaxial sites.36 For instance, the phenyl ring in Phe coincides almost perfectly with the hexagonal pattern of epitaxial sites on the Fe surface and as such makes a higher contribution to the adsorption strength than the aliphatic Met molecule. This is in agreement with the trend of our computed binding energies. The slightly higher experimental efficiency of Met is possibly due to additional covalent binding interactions (via the S atom of the thiol group), which were not captured in the computation. Nonetheless, it is clear that the almost perfect alignment of polarizable atoms in the molecules with epitaxial sites on the metal lattice accounts for the high physisorption strengths of the Fe–Phe and Fe–Met systems.
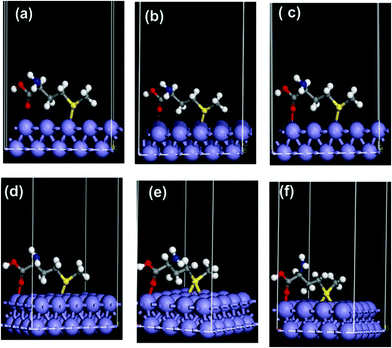 | ||
| Fig. 7 Perspective view of representative snapshots showing changes in adsorption orientation during chemisorption of Met on Fe (110), showing in different stages, the formation of Fe–S, Fe–O and S–Fe–S covalent bonds. The structures become energetically more favorable going from (a) to (e) with energy difference of 87.9 kcal mol−1. | ||
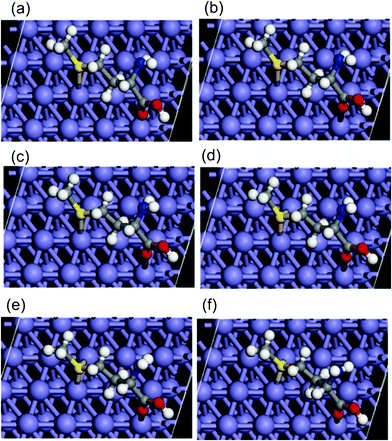 | ||
| Fig. 8 On-top view of representative snapshots showing changes in adsorption orientation during chemisorption of Met on Fe (110). The images show that the epitaxial adsorption orientation is retained during covalent interactions. | ||
Soft epitaxial adsorption is characterized by the spatially distributed nature of the dispersion interactions,38 and this requires relatively even surfaces to maintain the perfect fit of the molecular backbone of the adsorbate to the hexagonal pattern of epitaxial sites on the Fe surface. One should thus expect significant perturbation of this pattern as the metal surface becomes increasingly irregular with nanoprocessing, inducing significant lattice mismatch. The epitaxial adsorption process is thus disrupted, diminishing the degree of surface coverage and hence inhibition efficiency as observed for Phe on the nanocrystalline Fe surface. Since adsorption of Met also predominantly proceeds via the soft epitaxial mechanism, one should anticipate a similar scenario for Met on nanocrystalline Fe. However, the tendency of Met to interact covalently with the Fe surface needs to be taken into consideration.
An essential feature of covalent adsorption is the localized nature of the interactions, which means that such adsorption can occur favorably on locally even as well as locally uneven surfaces. Moreover, bonding of unsaturated organic molecules on noble metal surfaces has been shown to scale up significantly around defect sites on the surface,23–25,41 which implies that the high defect populations of nanocrystalline surfaces provide an abundance of active sites for bonding interactions with adsorbates with suitable electronic structures. The increased efficiency of Met on nanocrystalline Fe can thus be related to the ability of the Met molecule to interact covalently with the Fe surface and subsequent scaling up of such bonding interactions around the abundant defect sites on the nanocrystalline surface.
4. Experimental details
The test specimen was a conventional polycrystalline ingot iron, from which a bulk nanocrystalline ingot iron was prepared by severe rolling. The severe rolling technique and subsequent microstructure characterization of the polycrystalline and nanocrystalline Fe have been described in detail elsewhere.27 The specimens were machined into test coupons of dimension 1 cm × 1 cm, which were wet-polished with silicon carbide abrasive paper (from grade #600 to #1000), degreased in acetone, rinsed with distilled water and dried in warm air.28 The test corrodent was 0.5 M H2SO4, while the test inhibitors were methionine (Met) and phenylalanine (Phe) sourced from SCRC, China.Our experimental approach involved assessment of the potentiodynamic polarization and impedance behaviour of the metal specimens in the corrodent without and with 0.005 M concentrations of the test inhibitors using an AUTOLAB PGSTAT 30 potentiostat/galvanostat (Eco Chemie BV, Netherlands) equipped with General Purpose Electrochemical System (GPES 4.9) and Frequency Response Analyser (FRA) software, version 4.9. Test coupons of 1 cm2 exposed surface area were used as working electrodes, the counter and reference electrodes were a Pt foil and a saturated calomel electrode (SCE) respectively. The working electrode was immersed in a test solution for 1 h to attain a stable open circuit potential prior to each measurement. Potentiodynamic polarization studies were carried out in the potential range ±250 mV versus corrosion potential (Ecorr) at a scan rate of 0.333 mV s−1. Polarization resistance (Rp) was determined from the polarization curves in the vicinity of Ecorr (±10 mV). Electrochemical impedance spectroscopy (EIS) measurements were made at corrosion potentials (Ecorr) over a frequency range of 100 kHz–10 mHz, with a signal amplitude perturbation of 5 mV. All experiments were undertaken in stagnant aerated solutions at 30 ± 1 °C.
5. Conclusions
In summary, electrochemical measurements and theoretical calculations were employed to investigate the corrosion inhibition and adsorption behavior of phenylalanine (Phe) and methionine (Met) on polycrystalline and nanocrystalline iron. Experimental results reveal that Met was generally a better corrosion inhibitor than Phe. We also observed that the nanocrystalline Fe surface favored adsorption of Met, but diminished adsorption of Phe. We performed computational studies in the framework of the density functional theory in order to obtain molecular level information regarding the corrosion inhibition performance of Met and Phe on polycrystalline Fe and to account for the experimentally observed variations in the performance of both molecules on the nanocrystalline surface. No clear cut deductions could be drawn from quantum chemical parameters describing the electronic structures of the inhibitor molecules as the values are about the same for both molecules. The only significant difference was the remarkably high Fukui f− values associated with the S atom of the Met thiol group.The physisorption geometries of both molecules on Fe show clear evidence of soft epitaxial adsorption and the magnitudes of the physisorption energies agree more or less with the trend of experimentally determined inhibition efficiencies of Phe and Met on polycrystalline Fe, suggesting that soft epitaxial adsorption is the predominant effect controlling the corrosion inhibition performance of both molecules on this surface. Our computations also revealed an added ability of Met to interact covalently with Fe (via both the thiol S atom and the carbonyl O atom), which accounts for its slightly higher experimental inhibition efficiency on polycrystalline Fe.
Our findings show that the effect of metal microstructure modification on corrosion inhibitor adsorption depends ab initio on the nature of the interaction with the polycrystalline metal surface. Increased surface roughness and defect concentration associated with the nanocrystalline Fe surface perturbed the soft epitaxial adsorption patterns, with the ensuing lattice mismatch diminishing the strength of the physisorptive interactions. Fe surface nanocrystallization however promoted surface–molecule covalent interactions. Experimental evidence for both effects derives from the fact that inhibition efficiency of Met was higher on nanocrystalline Fe, while that of Phe was much reduced.
Acknowledgements
E.E. Oguzie is a TWAS-UNESCO Associate. Financial support from TWAS, the Academy of Sciences for the developing World, under the TWAS Grants for Research Units in Developing Countries Program (TWAS-RGA08-005) and from the National Natural Science Foundation of China under contract nos. 50501023 and 50771098 is gratefully acknowledged.References
- B. G. Ateya, B. E. El-Anadouli and F. M. A. El-Nizamy, Bull. Chem. Soc. Jpn., 1981, 54, 3157–3161 CrossRef CAS.
- M. A. Ameer, E. Khamis and G. Al-Senani, Adsorpt. Sci. Technol., 2000, 18, 177–193 CrossRef CAS.
- S. M. A. Shibli and V. S. Saji, Corros. Sci., 2005, 47, 2213–2224 CrossRef CAS.
- L. Tang, G. Mu and G. Liu, Corros. Sci., 2003, 45, 2251–2262 CrossRef CAS.
- P. Cao, R. Gu and Z. Tian, Langmuir, 2002, 18, 7609–7615 CrossRef CAS.
- D. W. Deberry, G. R. Peyton and W. S. Clark, Corrosion, 1981, 40, 250–256 CrossRef.
- Y. Harek and L. Larabi, Kem. Ind., 2004, 53, 55–61 CAS.
- M. Kliskic, J. Radosevic, S. Gudic and V. Katalinic, J. Appl. Electrochem., 2000, 30, 823–830 CrossRef CAS.
- K. F. Khaled, Electrochim. Acta, 2008, 53, 3484–3492 CrossRef CAS.
- G. Gece, Corros. Sci., 2008, 50, 2981–2992 CrossRef CAS.
- M. J. Bahrami, S. M. A. Hosseini and P. Pilvar, Corros. Sci., 2010, 52, 2793–2803 CrossRef CAS.
- J. Cruz, T. Pandiyan, E. Garcıa-Ochoa and J. Electroanal, Chem., 2005, 583, 8–16 CAS.
- J. Bartley, N. Huynh, S. E. Bottle, H. Flitt, T. Notoya and D. P. Schweinsberg, Corros. Sci., 2003, 45, 81–96 CrossRef.
- S. Martinez and I. S. Stagljar, THEOCHEM, 2003, 640, 167–174 CrossRef CAS.
- K. F. Khaled, K. Babic-Samardzija and N. Hackerman, Electrochim. Acta, 2005, 50, 2515–2520 CrossRef CAS.
- L. M. Rodrıguez-Valdez, A. Martınez-Villafane and D. Glossman-Mitnik, THEOCHEM, 2005, 713, 65–70 CrossRef.
- D. Turcio-Ortega, T. Pandiyan, J. Cruz and E. Garcia-Ochoa, J. Phys. Chem. C, 2007, 111, 9853–9866 CAS.
- M. Lashkari and M. R. Arshadi, Chem. Phys., 2004, 299, 131–137 CrossRef CAS.
- J. M. Roque, T. Pandiyan, J. Cruz and E. Garcıa-Ochoa, Corros. Sci., 2008, 50, 614–624 CrossRef CAS.
- Y. Duda, R. Govea-Rueda, M. Galicia and H. Beltran, J. Phys. Chem. B, 2005, 109, 22674–22684 CrossRef CAS.
- K. F. Khaled, Electrochim. Acta, 2009, 54, 4345–4352 CrossRef CAS.
- J. Fu, S. Li, Y. Wang, L. Cao and L. Lu, J. Mater. Sci., 2010, 45, 6255–6265 CrossRef CAS.
- A. Kokalj and S. Peljhan, Langmuir, 2010, 26, 14582–14593 CrossRef CAS.
- A. Kokalj, S. Peljhan, M. Finsgar and I. Milosev, J. Am. Chem. Soc., 2010, 132, 16657–16668 CrossRef CAS.
- K. J. Klabunde and R. S. Mulukutla, Nanoscale Materials in Chemistry, p. 223, Wiley, New York, 2001 Search PubMed.
- L. Li, Y. Li and F. Wang, J. Mater. Sci. Technol., 2010, 26, 1–14 Search PubMed.
- M. A. Johnson, Nat. Chem., 2009, 1, 8–9 CrossRef CAS.
- S. G. Wang, C. B. Shen, K. Long, H. Y. Yang, F. H. Wang and Z. D. Zhang, J. Phys. Chem. B, 2005, 109, 2499–2503 CrossRef CAS.
- E. E. Oguzie, S. G. Wang, Y. Li and F. H. Wang, J. Phys. Chem. C, 2009, 113, 8420–8429 CAS.
- B. J. Delley, Chem. Phys, 1990, 92, 508–518 CAS.
- B. J. Delley, Chem. Phys., 2000, 113, 7756–7765 CAS.
- C. J. Casewit, K. S. Colwell and A. K. Rappé, J. Am. Chem. Soc., 1992, 114, 10035–10046 CrossRef CAS.
- C. J. Casewit, K. S. Colwell and A. K. Rappé, J. Am. Chem. Soc., 1992, 114, 10046–10053 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- B. Delley, Phys. Rev. B: Condens. Matter, 2002, 66, 155125–155133 CrossRef.
- H. Heinz, B. L. Farmer, R. B. Pandey, J. M. Slocik, S. S. Patnaik, R. Pachter and R. R. Naik, J. Am. Chem. Soc., 2009, 131, 9704–9714 CrossRef CAS.
- M. Hoefling, F. Iori, S. Corni and K. Gottschalk, Langmuir, 2010, 26, 8347–8351 CrossRef CAS.
- J. Feng, R. B. Pandey, R. J. Berry, B. L. Farmer, R. R. Naik and H. Heinz, Soft Matter, 2011, 7(5), 2113 RSC.
- L. M. Ghiringhelli and L. Delle Site, J. Am. Chem. Soc., 2008, 130, 2634–2638 CrossRef CAS.
- S. B. Fard and M. Guagliano, Frattura ed Integrita Strutturale, 2009, 7, 3–16 Search PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |