Microstructure tuning of mesoporous silica prepared by evaporation-induced self-assembly processes: interactions among solvent evaporation, micelle formation/packing and sol condensation
Ying-Feng
Lee
a,
Kuo-Hsin
Chang
ab,
Che-Yi
Chu
a,
Hsin-Lung
Chen
a and
Chi-Chang
Hu
*a
aDepartment of Chemical Engineering, National Tsing Hua University, Hsin-Chu, 30013, Taiwan. E-mail: cchu@che.nthu.edu.tw; Fax: +886-3-5715408; Tel: +886-3-5736027
bDepartment of Chemical Engineering, National Chung Cheng University, Chia-Yi, 62102, Taiwan
First published on 9th August 2011
Abstract
By varying the vapor pressure of solvents through changing the alkyl length of aliphatic alcohols or the aging temperature, the microstructures, including mesopore ordering length, porosity, and specific surface area, of silica templates are controllable in an evaporation-induced self-assembly (EISA) process. The microstructures of various silica powders are systematically characterized and compared by the small-angle X-ray scattering (SAXS), nitrogen adsorption/desorption isotherms, as well as scanning electron microscopic (SEM) and transmission electron microscopic (TEM) images. The microstructure ordering length of silica powders is found to decrease with reduction of the solvent evaporation rate tuned by the length of alkyl groups of alcohols, which is confirmed by the almost identical microstructure of silica powders prepared by using methanol and tetrahydrofuran (THF) which exhibit similar vapor pressures. The rate of micelle formation relative to the rate of silica precursor condensation, strongly depending on the solvent evaporation rate, determines the resultant organization of micelles and may cause morphological distortion and microdomain disorientation of silica templates. This concept is confirmed by the presence of completely and partially ordered microstructures of silica powders synthesized from the ethanol- and butanol-based precursor solutions, respectively, which are dried completely at a higher temperature in the aging process. The microstructure of mesoporous silica can thus be simply tuned by varying solvents and aging temperatures in the EISA process, which are promising for many applications.
1. Introduction
Recently, there has been a significant breakthrough in the synthesis of open-framework inorganic materials of well-defined geometries, such as titania, carbons, silica, and so on.1–13 These kind of materials, generally synthesized by using self-assembled surfactants as templates, have attracted considerable interest because of their porous structures and high surface areas. These materials also show uniform pore size distributions and periodic porous microstructures. Among these porous materials, the development of nanostructured silica with precise pore shape and size control is of great importance in several fields of modern science and technology.14,15 These mesostructures are best appreciated in the systems where molecular recognition is needed; e.g., shape-selective catalysis, selective adsorption/separation processes, chemical sensors, molecular sieves, energy storage/conversion systems, as well as dielectric coatings of low dielectric constant materials.6–21To advance the applicability of these materials, the evaporation-induced self-assembly (EISA) process has been studied for many envisioned nanotechnologies. This unique method has been used to prepare most of such mesoporous materials. In fact, it is desirable to create patterned nanocomposites consisting of periodic arrangements of two or more dissimilar materials via this process. The above viewpoints imply that EISA is a simple, effective method for preparing periodic porous materials.22–24
EISA generally involves three interactive steps: solvent evaporation, micelle formation and packing, as well as sol condensation. The evaporation rate of solvents in the sol solution during the synthesis process is expected to be one of the key factors controlling the resultant microstructures in the EISA method because the surfactant concentration in the sol solution is strongly dependent upon the solvent evaporation rate. This may cause variations in structure and packing of micelles although ethanol is the most popular solvent for the EISA process. Accordingly, Innocenzi et al. gave an overall formation mechanism of ordered-mesoporous silica by using solvents with fast evaporation rates,25 and Ogura et al. described the so-called drying-induced phase transformation mechanism for the mesoporous silica formation.26 Recently, an ordered porous structure of silica could even be obtained through a so-called spray drying method,27 a rapid evaporation process (2 s) at elevated temperatures, further emphasizing the importance of the solvent evaporation rate. On the other hand, the rate of sol condensation is also important in determining the silica microstructure since condensation, very similar to polymerization, generally enhances the viscosity of a sol, which should significantly affect the formation and organization of micelles. Therefore, Doshi et al. showed that under acidic conditions in which the siloxane condensation was minimized, the hydrophilic and nonvolatile silicic acid components replaced water to maintain a fluid-like state that avoided kinetic barriers to self-assembly. This means that the condensation rate played an important role in forming the highly ordered surfactant-templated mesostructure.28 Recently, Choi et al. tried to replace the commonly used ethanol with 1-butanol and found the key synthetic strategy for achieving a robust, structurally well-ordered materials,29 further supporting the above viewpoint. Besides, due to the variation in length of alkyl groups and polarity of solvents, the controllable hydrophobicity of solvents may lead the microphase separation or templating of micelles in the EISA process, changing the porosity and microstructure of resultant materials. Based on these viewpoints, the interactions among solvent evaporation, micelle formation/packing, and sol condensation have to be carefully considered in preparing ordered mesoporous materials through the EISA process. Unfortunately, few researches considered these factors simultaneously in order to establish a reliable model for describing this process.
In this work, we vary the rate of solvent evaporation by changing the alkyl length of solvents, or by drying the sols at a relatively higher temperature (40 °C for 72 h) in order to figure out the competition among micelle formation, micelle packing, and gel network formation in the EISA process. This work concludes that the rate of micelle formation relative to the rate of silica precursor condensation, strongly depending on the solvent evaporation rate, determines the resultant organization of micelles and causes the morphological distortion and/or microdomain disorientation of silica templates. Moreover, the porous microstructure and ordering length of mesoporous silica can be simply tuned by varying the vapor pressure of solvents (i.e., the alkyl length of alcohols or the aging temperature) in the EISA process, which is promising for many future applications.
2. Experimental
2.1 Synthesis of mesoporous silica
Pluronic F127 (EO106PO70EO106) was dissolved in solvent and 12 M HCl was dropped in the above solution under stirring at 26 °C for 30 min. Then, tetraethyl-ortho-silicate (TEOS) was added into the above mixture under stirring for 30 min. The weight ratio of TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F127:HCl:water:solvent in the sol solution is 10:4.65:0.0615:2.6:4. Normally, the above sol mixtures in an open vessel were aged at 26 °C for 24 h to carry out the self-assembly process. However, two specified sols employing ethanol and n-butanol as the solvents were dried at 40 °C for 72 h to demonstrate the competition mechanism between micelle formation and silica gel condensation in the EISA process. All the aged sols were moved to an open-ended quartz tube in an oven, heated at a ramping rate of 1 °C min−1 to 450 °C, and kept at this temperature for 4 h to obtain the silica powders. In this work, five solvents, including methanol, ethanol, n-propanol, n-butanol, and THF, were tested and the resultant silica powders are denoted as SiO2-M, SiO2-E, SiO2-P, SiO2-B, and SiO2-T, respectively when the sols were normally dried at 26 °C for 24 h. On the other hand, the silica powders prepared from the ethanol- and n-butanol-based sols aged at 40 °C for 72 h are denoted as SiO2-EA and SiO2-BA, respectively.
:F127:HCl:water:solvent in the sol solution is 10:4.65:0.0615:2.6:4. Normally, the above sol mixtures in an open vessel were aged at 26 °C for 24 h to carry out the self-assembly process. However, two specified sols employing ethanol and n-butanol as the solvents were dried at 40 °C for 72 h to demonstrate the competition mechanism between micelle formation and silica gel condensation in the EISA process. All the aged sols were moved to an open-ended quartz tube in an oven, heated at a ramping rate of 1 °C min−1 to 450 °C, and kept at this temperature for 4 h to obtain the silica powders. In this work, five solvents, including methanol, ethanol, n-propanol, n-butanol, and THF, were tested and the resultant silica powders are denoted as SiO2-M, SiO2-E, SiO2-P, SiO2-B, and SiO2-T, respectively when the sols were normally dried at 26 °C for 24 h. On the other hand, the silica powders prepared from the ethanol- and n-butanol-based sols aged at 40 °C for 72 h are denoted as SiO2-EA and SiO2-BA, respectively.
2.2 Characterization
The BET surface area and average BJH pore size of silica powders were obtained from N2adsorption/desorption analysis at 77 K (Autosorb-1, Quantachrome). The BET surface areas were calculated from the adsorption isotherms in the relative pressure range from 0.025 to 0.3. The average pore diameter was estimated by means of the BJH model from the desorption isotherms, while for ordered SiO2-M and SiO2-T, the average pore diameter was calculated from the adsorption isotherms due to the tensile strength effect on the cylindrical meniscus.30 The micropore diameter were estimated from the DA method.31 The mesopore and micropore volumes were obtained from the V–T plot.32 The total pore volume was derived from the amount of vapor adsorbed at a relative pressure close to unity.32 The morphologies and microstructures of samples were characterized by high-resolution transmission electron microscopy (HR-TEM) using JEOL 2100 or Philips FEI-TEM microscopes. Field-emission scanning electron microscopic (FESEM) observations were performed on a JEOL JSM-6700F using accelerating voltages of 3.0 keV.3. Results and discussion
3.1 Effects of the alkyl length of solvents
Fig. 1 shows the small angle X-ray scattering (SAXS) patterns of various silica powders prepared in this work. The SAXS profile of SiO2-M (curve 1) reveals the hexagonal-packed cylinder phase as a series of diffraction peaks with the position ratio of 1:31/2:41/2 (red arrows) are identified. From a detailed comparison of all SAXS patterns, the presence of a board peak, an undefined peak, and a monotonously decayed curve for SiO2-E, SiO2-P, and SiO2-B, respectively, indicates a more disordered microstructure when the alkyl length of the solvent becomes longer. Since the vapor pressure (and hence solvent evaporation rate) monotonously decrease with the length of alkyl groups for the alcohols investigated here, the observed solvent dependence of disordering of silica templates should be influenced by the solvent evaporation rate although longer alkyl length of alcohols may hinder the formation of a long-range ordering pore structure, leading to pore structure collapse. Clearly, the solvent with a sufficiently fast evaporation rate (i.e., methanol) will accelerate the concentration of surfactants, favoring the formation of cylindrical micelles. These micelles should organize spontaneously into the hexagonal lattice if packing is not significantly inhibited by the minor gel condensation. The formation of cylinder micelles with certain distortion is possible when a solvent with a slight decrease in the evaporation rate is employed. This incomplete organization of micelles will result in morphological distortion and surfactant microdomain disorientation (e.g., SiO2-E and SiO2-P) because relatively significant gel condensation will depress the self-organization of micelles. When a solvent with a very low evaporation rate is employed in the precursor solution, the concentration of surfactant may not reach the critical micelle concentration (CMC). Accordingly, formation and organization of micelles becomes insignificant and a seriously distorted, macrophase-separated morphology with a random texture is obtained (i.e., SiO2-B).
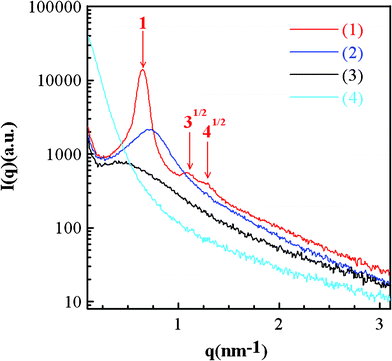 | ||
| Fig. 1 The small-angle X-ray scattering patterns of (1) SiO2-M (methanol with a vapor pressure = 97.6 mmHg at 20 °C), (2) SiO2-E (ethanol with a vapor pressure = 44.3 mmHg at 20 °C), (3) SiO2-P (n-propanol with a vapor pressure = 14.9 mmHg at 20 °C), and (4) SiO2-B (n-butanol with a vapor pressure = 3.75 mmHg at 20 °C).34 | ||
Identifying the pore structure, commonly determined by the adsorption/desorption isotherms of inert gases, is an essential procedure in template synthesis. Isotherms A–D in Fig. 2 show the typical adsorption/desorption isotherms of N2 at 77 K for SiO2-M, SiO2-E, SiO2-P, and SiO2-B, respectively. From a comparison of these isotherms, two features have to be mentioned. First, the order of silica with respect to decreasing the intercept value from the extrapolation of the adsorption/desorption overlapping lines in the low relative pressure region is: SiO2-E > SiO2-M > SiO2-P > SiO2-B, suggesting that the specific surface area of silica generally decreases with decreasing the vapor pressure of the alcohols. Second, hysteresis loops are found on all isotherms and all silica exhibit the type IV isotherms, typical for mesoporous materials. A feature common to hysteresis loops is that the steep region of the desorption branch leads to a lower closure point at a relative pressure (for a given adsorption). This phenomenon depends mainly on the nature of adsorbates rather than the nature of porous templates; e.g., for benzene at 25 °C at P/P0 = 0.28 and for N2 at its boiling point at P/P0 = 0.42. Note that SiO2-M shows a type H1 hysteresis loop, indicating a narrow distribution of cylindrical-like mesopores. This result reveals the formation of a uniform and highly ordered microstructure of SiO2-M. On the other hand, SiO2-E and SiO2-P exhibit a loop similar to type H2, suggesting a complex porous microstructure, such as network and pore blocking effects, and consequently, the pore size distribution and pore shape are not well defined. SiO2-B shows the type H3 hysteresis loop which was commonly found for the adsorbents with slit-shaped pores or plate-like particles.32 The above results further support the statement that the solvent with a high enough evaporation rate (i.e., methanol) favors the formation of ordered cylinders arranged in a hexagonal-like array.
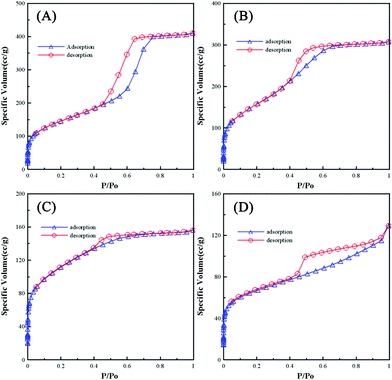 | ||
| Fig. 2 Nitrogen adsorption/desorption isotherms of (A) SiO2-M, (B) SiO2-E, (C) SiO2-P, and (D) SiO2-B. | ||
Table 1 summarises the physical properties of all silica. The specific surface area data were deduced from the adsorption isotherms on the basis of the BET model. From Table 1, SiO2-M possesses a high specific surface area because the ordered silica structure will induce a high packing arrangement. Due to the compact structure, the micropore diameter of SiO2-M is also the largest among these samples. SiO2-E shows the highest specific surface area because the slight shrinkage/collapse of the ordered structure leads to the formation of defect pores providing more surface area than SiO2-M. Since the average diameters of micropores and mesopores of SiO2-E are significantly smaller than that of SiO2-M, the former silica with smaller micropore and mesopore volumes still shows a higher specific surface area (561.37 m2 g−1) than SiO2-M. SiO2-P is considered as a micropore-enriched mesoporous silica although its micropore volume is not very high. Since SiO2-B shows the macrophase-separated morphology with a random texture, the silica precursors should aggregate seriously during the EISA process, generating certain larger pores in the template, and significant amount of mesopores is obtained. Based on the above results and discussion, the morphology and pore structure of silica can be tuned by varying the solvent evaporation rate through varying the length of alkyl group in the EISA process.
| Sample code | SiO2-M | SiO2-E | SiO2-P | SiO2-B | SiO2-T |
|---|---|---|---|---|---|
| Surface area (m2 g−1) | 511 | 561 | 387 | 226 | 462 |
| Total pore volume (cc g−1) | 0.634 | 0.467 | 0.241 | 0.200 | 0.528 |
| BJH pore diameter (nm) | 6.53 | 3.43 | 3.38 | 3.76 | 5.63 |
| Micropore diameter (nm) (DA method) | 1.88 | 1.64 | 1.70 | 1.64 | 1.84 |
| Mesopore volume (cc g−1) (V–T plot) | 0.581 | 0.418 | 0.211 | 0.095 | 0.483 |
| Micropore volume (cc g−1) (V–T plot) | 0.026 | 0.034 | 0.017 | 0.013 | 0.023 |
The FE-SEM image in Fig. 3A clearly shows that SiO2-M is of an ordered hexagonal structure and the pore size is about 6 nm, revealing the mesoporous nature. Due to the difference in solvent evaporation rates that results in the distortion of the micelle microstructure, SiO2-E and SiO2-P show many pore-like domains with relatively uniform micropores. SiO2-E shows a higher density of pores with their sizes less than 2 nm, significantly enhancing the specific surface area (see Table 1). SiO2-P exhibits more loose pores with their pore size generally smaller than 2 nm (i.e., micropores). SiO2-B seems to show the aggregated-shape plate morphology, suggesting the macrophase separation between solvent and triblock copolymer (i.e., Pluronic F127). The above results are consistent with those of nitrogen adsorption/desorption isotherms and SAXS patterns, further supporting the idea that the solvent evaporation rate in the sol solution during the EISA synthesis process is, at least, one of the key factors controlling the resultant microstructures of the silica.
 | ||
| Fig. 3 FE-SEM images of (A) SiO2-M, (B) SiO2-E, (C) SiO2-P, and (D) SiO2-B. | ||
To further confirm the variation in the morphologies of mesoporous silica formed under different evaporation rates of solvents, all samples were examined by TEM and typical results are shown in Fig. 4. Clearly, SiO2-M shows an ordered hexagonal structure. SiO2-E and SiO2-P can be considered as porous materials providing random pores. For SiO2-B, large aggregates of silica are clearly visible, resulting from the macrophase separation between n-butanol and Pluronic F127. Based on all of the above results and discussion, the microstructure of silica synthesized via the EISA process strongly depends on the evaporation rate (vapor pressure) of solvent employed.
 | ||
| Fig. 4 TEM images of (A) SiO2-M, (B) SiO2-E, (C) SiO2-P, and (D) SiO2-B. | ||
3.2 Effects of solvent polarity
Due to the fact that different solvents may show similar vapor pressures, resulting in a similar solvent evaporation rate, the microstructures of silica templates prepared from the EISA process with methanol and THF (a solvent with weak polarity33 and a high vapor pressure34) are compared in this work. In Fig. 5A, the SAXS pattern of SiO2-T shows a characteristic peak associated with an ordered hexagonal phase (blue arrows). This result reveals that this weakly polar solvent with a vapor pressure close to methanol shows the same effect as methanol on the microstructure of the silica powders. This statement is supported by the N2 adsorption/desorption isotherms in Fig. 5B which exhibits a type H1 hysteresis loop, indicating a uniform and highly ordered microstructure of SiO2-T. From Table 1, the average diameters of micropores and mesopores of SiO2-T are very close to that of SiO2-M, indicating the very similar pore structure between these two silica powders. In addition, the SAXS diffraction peaks with the position ratio of 1:31/2:41/2 (Fig. 5A) are more obvious from a comparison of the SAXS pattern in Fig. 5A and curve 1 in Fig. 1A, indicating a longer range order structure of SiO2-T. The explanation is further confirmed by the pore analysis, since SiO2-T shows a smaller specific surface area (462.1 m2 g−1) than SiO2-M (511.12 m2 g−1). The above result suggests that packing of micelles in the sol solution during the EISA synthesis process with THF as the solvent is denser than that with methanol as the solvent. On the other hand, a highly ordered hexagonal structure of SiO2-T is clearly found in Fig. 5C, reveals the highly similar microstructures between SiO2-M and SiO2-T. Based on the above similarities in the SAXS patterns, TEM images, and BET pore structures of SiO2-M and SiO2-T, we conclude that the evaporation rate of solvents (and vapor pressure) is a key factor in determining the resultant microstructure of silica templates in the EISA process.
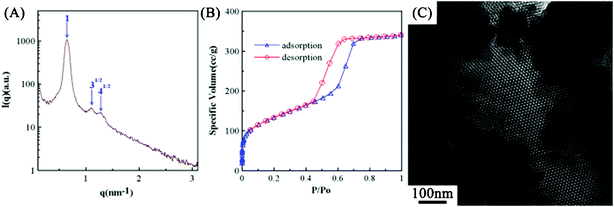 | ||
| Fig. 5 (A) The small-angle X-ray scattering pattern, (B) the nitrogen adsorption/desorption isotherms, and (C) the TEM image of SiO2-T prepared from the sol solution with THF as the solvent. | ||
3.3 Effects of the aging process
Based on the foregoing and experimental results, there is a doubt that the macrophase separation between n-butanol and Pluronic F127 is induced by the formation of viscous silica gel network during the heating process at a ramping rate of 1 °C min−1 to 450 °C because solvents, especially n-butanol, were not effectively evaporated during the 24 h, room-temperature, aging process. This incomplete evaporation of solvents will result in the variation of surfactant concentration, probably leading to ineffective formation of micelles or poor packing of the self-assembled micelles. Consequently, the following heating process, favorable to the formation of viscous silica gel network, will inhibit the surfactant movement and micelle packing. This idea is tested by employing SiO2-EA and SiO2-BA whose respective ethanol-based and n-butanol-based sol solutions were (nearly) completely dried at 40 °C for 72 h to demonstrate the competition between micelle formation and silica gel condensation in this EISA process. Note in Fig. 6A and 6C that both SAXS patterns demonstrate the highly ordered structure of SiO2-EA and SiO2-BA as evidenced from the ratio of diffraction peak positions of 1:31/2:71/2 (orange arrows). In Fig. 6B, a periodically ordered hexagonal structure is clearly obtained although SiO2-E (see Fig. 4B) does not show such an excellent, highly long-range ordered microstructure. Since most ethanol molecules were found to evaporate in a period much shorter than 24 h in the above 40 °C-drying process, the formation and packing of micelles should be fast and complete. Consequently, a highly periodic hexagonal structure is obtained. The above result strongly supports two important concepts. First, a long-range ordered microstructure of silica template can be obtained through effective formation and packing of micelles before the significant formation of silica gel. Second, the pore ordering structure of silica can be tuned by varying the solvent evaporation rate through varying the aging temperature in the EISA process. On the other hand, a mixed microstructure consisting of an ordered hexagonal structure and aggregated silica is obtained when n-butanol is employed as the solvent (see Fig. 6D). This phenomenon is attributable to the requirement of 72 h drying in order to carry out the nearly complete evaporation of n-butanol, confirming the competition mechanism between micelle formation and silica gel condensation in the EISA process. The much longer aging time at 40 °C should result in the significant micelle formation/packing. However, gel condensation should occur significantly before the complete formation and packing of micelles due to the above long aging time. The above competition leads to a partially long-range ordered silica gel network of SiO2-BA. A similar finding on the change of ordering structure by varying the aging temperature has also been reported by Yang and co-worker,35,36 further supporting our postulate.
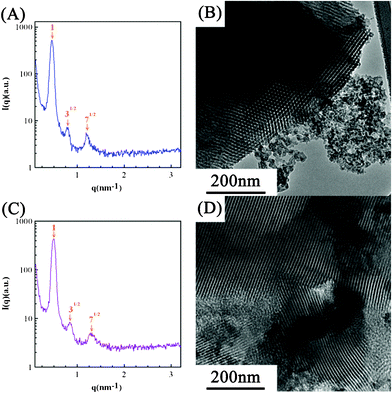 | ||
| Fig. 6 (A,C) The small-angle X-ray scattering patterns and (B,D) TEM images of (A,B) SiO2-EA and (C,D) SiO2-BA where the sol solutions were dried at 40 °C for 72 h. | ||
Based on all of the above results and discussion, we propose a scheme illustrating the complicated interactions among micelle formation, micelle packing, and gel network formation in the EISA process (see Scheme 1). Because of the amphiphilic characteristics of the PEO–PPO–PEO triblock copolymer, Pluronic F127 can form spherical, cylinder, and hexagonal-packed micelles due to its self-assembling nature in the sol solution, which strongly depends on the surfactant concentration. When a solvent with a sufficient vapor pressure (e.g., methanol and THF) is employed, the solvent evaporation rate is high and Pluronic F127 is concentrated to form cylindrical micelles before the formation of a viscous silica gel network. Under this situation, the self-assembly process is fast enough to pack into the ordered hexagonal phase before the application of the temperature-ramping program. Consequently, a highly ordered hexagonal structure is obtained for SiO2-M and SiO2-T (path 1). Similarly, if solvent molecules are completely evaporated in a much shorter period by rising the aging temperature before the significant formation of viscous silica gel network, concentrated Pluronic F127 molecules will form the cylindrical micelles which pack into an ordered hexagonal phase. Consequently, a highly ordered hexagonal structure is obtained (e.g., SiO2-EA synthesized from the completely dried sol solution in Fig. 6A and 6B). This aging step shows the potential in finely tuning the microstructure ordering length, porosity, and specific surface area. When n-butanol is employed, the aged sol cannot be dried at 26 °C for 24 h, which will be dried completely by the temperature-ramping program. However, the temperature-ramping program not only favors the solvent evaporation but also accelerates the condensation of TEOS to form silica. Accordingly, micelles may only be formed in certain places while the viscous silica gel aggregates when the sol–gel condensation is significant (path 2). Hence, silica powders with a macrophase-separated morphology with a random texture are obtained (i.e., SiO2-B). Since the vapor pressures of ethanol and n-propanol are between methanol and n-butanol, the resultant microstructures of silica are intermediate to the periodically ordered and macrophase-separated morphologies. Since the evaporation rate of n-propanol is relatively slow, not well-defined micelles may be formed in the aged process. Hence, it is impossible to pack these ill defined-micelles into an ordered microstructure (e.g., SiO2-P). Moreover, the microstructure of SiO2-E should be a hybrid of SiO2-M and SiO2-P because the vapor pressure and evaporation rate of ethanol are between methanol and n-propanol. On the other hand, if solvent molecules with a low vapor pressure are (nearly) completely evaporated by prolonging the aging time (path 3), such as n-butanol with a relative long alkyl chain, the condensation of viscous silica gel network occurs significantly because of a much longer aging time. As a result, micelle formation/packing and gel condensation complete each other in this long aging period, leading to a mixed microstructure consisting of an ordered hexagonal structure and aggregated silica (i.e., SiO2-BA, see Fig. 6C and 6D).
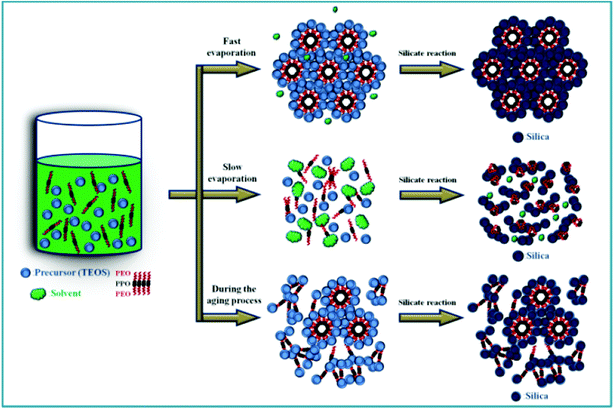 | ||
| Scheme 1 A schematic illustration describes how to control the degree of surfactant self-assembly by changing the relatively rates of solvent evaporation and gel formation in the EISA process. | ||
Based on all of the above results and discussion, the relative rates of micelle formation and TEOS condensation are concluded to determine the morphology and packing structure of micelles, resulting in the variation in morphologies and microstructures of mesoporous silica templates. Crucially, the degree of surfactant self-assembly, strongly depending on the solvent evaporation rate, can be used to control the micelle morphology and packing as well as the resultant microstructure of silica templates. In addition, the microstructure of silica templates can be tuned by varying the solvent with similar vapor pressures from the textural comparison of SiO2-M and SiO2-T or by shortening the aging time through rising the aging temperature from the textural comparison of SiO2-E and SiO2-EA.
4. Conclusions
According to the SAXS patterns, nitrogen adsorption/desorption isotherms, SEM images, and TEM images of silica templates prepared in this work, the ordered length of silica templates is monotonously decreased with decreasing the evaporation rate of solvent in the EISA process. The surfactant concentration in the sol solution, strongly depending on the solvent evaporation rate, determines the morphology and packing structure of micelles, effectively controlling the resultant microstructure of silica templates. The similarities in the microstructure of SiO2-M and SiO2-T as well as the similar vapor pressures of methanol and THF confirm the idea that the evaporation rate of solvents is the key factor determining the resultant microstructure of silica templates synthesized via the EISA process. The microstructures of mesoporous silica templates can be simply controlled by changing the relative rates of solvent evaporation and TEOS condensation in the aging step of the EISA process, which are promising for several future applications.Acknowledgements
The financial support of this work, by the National Science Council of ROC Taiwan under contract no. NSC 100-3113-E-006-001 and the boost program of NTHU, is gratefully acknowledged.References
- C. W. Wu, T. Ohsuna, M. Kuwabara and K. Kuroda, J. Am. Chem. Soc., 2006, 128, 4544 CrossRef CAS.
- S. Tominaka, C. W. Wu, T. Momma, K. Kuroda and T. Osaka, Chem. Commun., 2008,(25), 2888 RSC.
- D. Chen, L. Cao, F. Huang, P. Imperia, Y. B. Cheng and R. A. Caruso, J. Am. Chem. Soc., 2010, 132, 4438 CrossRef CAS.
- P. Behrens, Adv. Mater., 1993, 5, 127 CrossRef CAS.
- T. J. Barton, L. M. Bull, W. G. Klemperer, D. A. Loy, B. McEnaney, M. Misono, P. A. Monson, G. Pez, G. W. Scherer, J. C. Vartuli and O. M. Yaghi, Chem. Mater., 1999, 11, 2633 CrossRef CAS.
- A. K. Cheetham, G. Ferey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268 CrossRef CAS.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237 CrossRef CAS.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896 CrossRef CAS.
- S. Yang, X. Zhou, P. Yuan, M. Yu, S. Xie, J. Zou, G. Q. Lu and C. Yu, Angew. Chem., Int. Ed., 2007, 46, 8579 CrossRef CAS.
- J. Tang, X. Zhou, D. Zhao, G. Q. Lu, J. Zou and C. Yu, J. Am. Chem. Soc., 2007, 129, 9044 CrossRef CAS.
- B. P. Ladewig, R. B. Knott, A. J. Hill, J. D. Riches, J. W. White, D. J. Martin, J. C. D. Costa and G. Q. Lu, Chem. Mater., 2007, 19, 2372 CrossRef CAS.
- Y. F. Lee, K. H. Chang, C. C. Hu and K. M. Lin, J. Mater. Chem., 2010, 20, 5682 RSC.
- C. W. Wu, Y. Yamauchi, T. Ohsuna and K. Kuroda, J. Mater. Chem., 2006, 16, 3091 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- D. Zhao, P. Yang, N. Melosh, J. Feng, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1998, 10, 1380 CrossRef CAS.
- T. Yamada, H. S. Zhou, H. Uchida, M. Tomita, Y. Ueno, T. Ichino, I. Honma, K. Asai and T. Katsube, Adv. Mater., 2002, 14, 812 CrossRef CAS.
- H. Zhou, S. Zhu, M. Hibino, I. Honma and M. Ichihara, Adv. Mater., 2003, 15, 2107 CrossRef CAS.
- Y. Brian, H. S. Zhou, T. Yamada, K. Asai and I. Honma, ChemPhysChem, 2004, 5, 261 CrossRef.
- T. Y. Wei, S. Y. Lu and Y. C. Chang, J. Phys. Chem. B, 2008, 112, 11881 CrossRef CAS.
- T. Y. Wei, S. Y. Lu and Y. C. Chang, J. Phys. Chem. C, 2009, 113, 7424 CAS.
- G. J. A. A. Soler-Illia, A. Louis and C. Sanchez, Chem. Mater., 2002, 14, 750 CrossRef.
- D. Grosso, F. Cagnol, G. J. A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309 CrossRef CAS.
- D. Grosso, C. Boissiere, B. Smarsly, T. Brezesinski, N. Pinna, P. A. Albouy, H. Amenitsch, M. Antonietti and C. Sanchez, Nat. Mater., 2004, 3, 787 CrossRef CAS.
- P. Innocenzi, L. Malfatti, T. Kidchob and P. Falcaro, Chem. Mater., 2009, 21, 2555 CrossRef CAS.
- M. Ogura, H. Miyoshi, S. P. Naik and T. Okubo, J. Am. Chem. Soc., 2004, 126, 10937 CrossRef CAS.
- I. V. Melnyk, Y. L. Zub, E. Veron, D. Massiot, T. Cacciaguerra and B. Alonso, J. Mater. Chem., 2008, 18, 1368 RSC.
- D. A. Doshi, A. Gibaud, V. Goletto, M. Lu, H. Gerung, B. Ocko, S. M. Han and C. J. Brinker, J. Am. Chem. Soc., 2003, 125, 11646 CrossRef CAS.
- S. Y. Choi, M. Mamak, N. Coombs, N. Chopra and G. A. Ozin, Adv. Funct. Mater., 2004, 14, 335 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. P. Ramirez, Microporous Mesoporous Mater., 2003, 60, 1 CrossRef CAS.
- I. Tanahashi, A. Yoshida and A. Nishino, Carbon, 1991, 29, 1033 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201 CrossRef.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, New York, 2007 Search PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, 4th ed., J. Wiley & Sons, Inc., New York, 1999 Search PubMed.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 2813 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |