PEGylation of surface protein filaments: coverage and impact on denaturation†
Stacy
Slavin
a,
Ezat
Khoshdel
b and
David M.
Haddleton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, United Kingdom. E-mail: D.M.Haddleton@warwick.ac.uk; Tel: +44 24 7652 3256; Fax: +44 24 7652 4112
bUnilever Research, Port Sunlight, Bebington, CH63 3JW, United Kingdom
First published on 18th July 2011
Abstract
N-Hydroxysuccinimidyl functional polymers for multi-site bioconjugation to amine residues present in α-keratin protein filaments were synthesised via living radical polymerisation. Monomers containing di(ethylene glycol) and poly(ethylene glycol) were employed to give water soluble polymers which were characterised using 1H NMR and size exclusion chromatography (SEC). The denaturation temperature of the PEG–keratin conjugates was examined using differential scanning calorimetry (DSC) to show that after conjugation to damaged keratinous material, the structural properties of the protein were improved against thermal degradation. Scanning electron microscopy was used to visualise surface coverage.
Introduction
Polymer–protein conjugation is employed for application in different fields, from therapeutics to personal care with the aim of using water soluble polymers which can attach to proteins and change their structure while maintaining biological function and properties.1–3 Poly(ethylene glycol) (PEG) containing polymers are a popular choice for conjugation as they are relatively safe and approved for use in vivo and have high water solubility.4 Advances in polymer–protein conjugation are highlighted in several recent reviews and papers, covering the progress made since the first polymer–protein conjugation reactions which were reported by Abuchowski and co-workers in the 1970's.5–12Poly(ethylene glycol) methyl ether methacrylate(s) (PEGMEMA) polymerised from functional initiators is an established approach to ‘PEGylation’ chemistry.13–15 The ability to incorporate functionality at the end of the polymer chain allows for a precise and selective point of conjugation. In this present study an initiator containing N-hydroxysuccinimide is employed, allowing for conjugation to accessible amines present in proteins either as end-amines or lysine residues. Synthesis of these initiators has been reported previously and polymers initiated from N-succinimidyl-2-bromopropionate (a) (Fig. 1) are more reactive towards nucleophiles, than polymers initiated by N-succinimidyl-2-bromo-2-methylpropionate (b).15 This has also been observed by Roberts et al. for α-branched N-succinimidyl esters–mPEG; however, when synthesising these polymers it should be noted that although better conjugation reactivity is observed, poor initiator efficiency is seen for the polymerisation.2,16
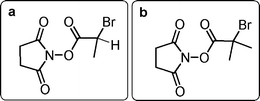 | ||
| Fig. 1 Structures of N-succinimidyl ester polymerisation initiators. | ||
Further initiators have been developed to conjugate polymers to proteins; ranging from the employment of thiol-ene click reactions to functional initiators bearing aldehyde and maleimide terminal groups.17–21 The approach used in this present study involves the synthesis of a preformed polymer for grafting to proteins. An alternative method is the ‘grafting from’ approach, where proteins are transformed to macroinitiators for subsequent polymerisation from the modified biological.22
Previously, Nicolas et al. have demonstrated direct conjugation of preformed polymers to hair.23 This confirmed that bioconjugation onto three dimensional complex biological surfaces can be carried out using conventional PEGylation chemistry. These polymerisations were carried out using N-succinimidyl-2-bromopropionate incorporating a fluorescent tag, hostasol (HMA), attached to enable demonstration of bioconjugation via confocal microscopy. This present work expands on these observations and focuses on a larger series of polymers investigating the effect of changing polymer chain length on the final polymer–protein properties.
Confocal images from our previous paper confirmed attachment of polymer to the hair surface, as opposed to internalisation, whilst differential scanning calorimetry (DSC) was carried out to measure the change in denaturation temperature of hair samples. DSC allows measurement of the denaturation temperature, Td, of damaged hair which differs from healthy hair, which has been used to confirm that the attached polymer has improved the thermal properties of damaged hair.24–25
Hair has a complex composition which can be considered to have three main constituents, each of which contains several levels of structural complexity (Scheme 1).26–27 Chemical reactions can take place on the surface of hair, at the cuticle and also on the cortex underneath, when it is exposed. The cuticle consists of overlapping scales which wrap around the cortex acting as a protective barrier while the central medulla is a cavity made up of cells of amorphous proteins.
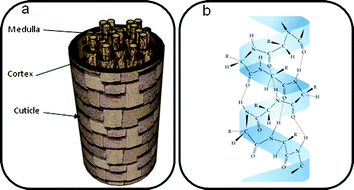 | ||
| Scheme 1 (a) The main components of hair and (b) α-keratin structure | ||
A review on the structure of hair by Crisan Popescu et al. provides an insight into the microscopic structure of hair. Hair is composed of 90–97% proteins which have amino acid residues on the surface available for conjugation. The fibrous protein α-keratin (Scheme 1) exists in hair as three α-helices twisted together (a protofibril), and these helices are coiled-coil and wrap around each other to form a ‘supercoil’ structure.28 Due to the primary structure of α-keratin, the surface of hair is negatively charged and can absorb cationic species readily onto the surface. Disulfide and isodipeptide bonds within the hair structure provide structural and mechanical strength.29–30 In particular, the ‘A-layer’ found in the cuticle has a high content of disulfide bonds which gives thermal stability to hair. Hair as a whole is hygroscopic and the water content within hair has a huge impact on its mechanical and thermal properties.24,31–33
Polymers can react with hair by physical interactions via deposition, adhesion, absorption or adsorption, or covalently by grafting.34–40 The choice of administration method depends on the desired properties of the hair. Non-covalent methods, such as deposition is popular, for example, in hair conditioners. The polymer is then held on the hair surface via electrostatic interactions. To carry out covalent polymer conjugation to hair, the polymer must be tailor-made to target a reactive site in hair. The conjugation approach we chose targeted lysine residues which have high surface area coverage of the cuticle of approximately 330 μmol g−1.27
Experimental methods
Materials
Copper(I) bromide (Aldrich, 98%) was purified according to the method of Keller and Wycoff.41N-(Ethyl)-2-pyridylmethanimine was prepared as described previously and stored at 0 °C under a dinitrogen atmosphere.42 Monomers DEGMEMA (Aldrich, 95%) and (PEGMEMA)475 (Aldrich, 95%) were used as received. Initiators were synthesised using methods previously established.15 All other reagents and solvents were obtained at the highest purity available from Aldrich Chemical Co. and used without further purification unless stated.Analytical methods
Polymerisations were carried out using standard Schlenk techniques under an inert atmosphere of nitrogen unless otherwise mentioned. Monomer conversions were calculated by 1H NMR by monitoring the disappearance of the peaks corresponding to the vinylic protons (5.6 and 6.2 ppm) with mesitylene (6.9 ppm) as an internal standard. Molar mass distributions were measured using size exclusion chromatography (SEC), on a system equipped with two PL gel 5 μm mixed D-columns (300 × 7.5 mm) and a PL gel 5 mm guard column (50 × 7.5 mm) (Polymer Laboratories) with differential refractive index detection using chloroform/triethylamine 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 at 1.0 mL min−1 as eluent. Poly(MMA) standards were used to calibrate the SEC. The analysed samples contained (0.2% vol) toluene as the flow marker. 1H NMR spectra were obtained on a Bruker DPX400 spectrometer. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane, referenced to the chemical shifts of residual solvent resonances. The molecular weight of the polymers Mn(NMR) are calculated by comparing the integrals of the chain-end signals and appropriate peaks from the polymer backbone. DSC experiments were carried out at Unilever, Port Sunlight using a Mettler Toledo DSC1 and STARe system software. SEM images were captured using a ZEISS SUPRA55VP.
:5 at 1.0 mL min−1 as eluent. Poly(MMA) standards were used to calibrate the SEC. The analysed samples contained (0.2% vol) toluene as the flow marker. 1H NMR spectra were obtained on a Bruker DPX400 spectrometer. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane, referenced to the chemical shifts of residual solvent resonances. The molecular weight of the polymers Mn(NMR) are calculated by comparing the integrals of the chain-end signals and appropriate peaks from the polymer backbone. DSC experiments were carried out at Unilever, Port Sunlight using a Mettler Toledo DSC1 and STARe system software. SEM images were captured using a ZEISS SUPRA55VP.
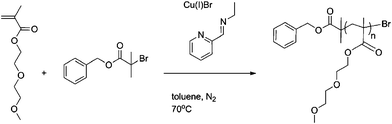
Polymer Series 1: Polymerisation of PEGMEMA monomer with control and reactive initiators
The dark green solution was subsequently heated to 70 °C with constant stirring. Samples were removed periodically using a degassed syringe for molecular weight (SEC) and conversion analysis (1H NMR). After 7 h, air was bubbled through the mixture for 0.5 h and the green suspension was kept at 0 °C overnight. After filtration through a short neutral alumina column, to remove the green copper complexes, toluene was removed with reduced pressure and the polymer dissolved in dichloromethane. The crude polymer solution was then purified by precipitation from petroleum ether 40/60 and remaining solvents were removed under vacuum. Mn(NMR) = 5000 g mol−1, Mw/Mn= 1.09. (Scheme 2.)
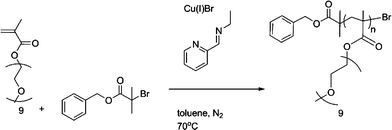 | ||
| Scheme 2 General reaction conditions for control Bnz-p(PEGMEMA). | ||
The dark green solution was subsequently heated to 70 °C with constant stirring. Samples were removed periodically using a degassed syringe for molecular weight (SEC) and conversion analysis (1H NMR). After 7 h, air was bubbled through the mixture for 0.5 h and the green suspension was kept at 0 °C overnight. After filtration through a short acidic alumina column, to remove the green copper complexes, toluene was removed with reduced pressure and the polymer dissolved in dichloromethane. The crude polymer solution was then purified by precipitation from petroleum ether 40/60 and remaining solvents were removed under vacuum. Mn(NMR) = 10 500 g mol−1, Mw/Mn = 1.09. (Scheme 3.)
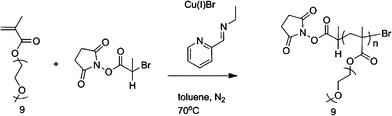 | ||
| Scheme 3 General reaction conditions for reactive NHS-p(PEGMEMA). | ||
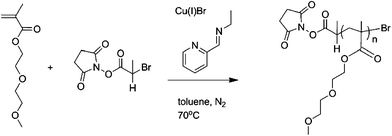
Polymer Series 2: Polymerisation of DEGMEMA monomer with control and reactive initiators
The dark green solution was subsequently heated to 70 °C with constant stirring. Samples were removed periodically using a degassed syringe for molecular weight (SEC) and conversion analysis (1H NMR). After 7 h, air was bubbled through the mixture for 0.5 h and the green suspension was kept at 0 °C overnight. After filtration through a short neutral alumina column, to remove the green copper complexes, toluene was removed with reduced pressure and the polymer dissolved in dichloromethane. The crude polymer solution was then purified by precipitation from petroleum ether 40/60 and remaining solvents were removed under vacuum. Mn(NMR)= 4900 g mol−1, Mw/Mn = 1.13. (Scheme 4.)
 | ||
| Scheme 4 General reaction conditions for control Bnz-p(DEGMEMA) | ||
The dark green solution was subsequently heated to 70 °C with constant stirring. Samples were removed periodically using a degassed syringe for molecular weight (SEC) and conversion analysis (1H NMR). After 7 h, air was bubbled through the mixture for 0.5 h and the green suspension was kept at 0 °C overnight. After filtration through a short acidic alumina column, to remove the green copper complexes, toluene was removed with reduced pressure and the polymer dissolved in dichloromethane. The crude polymer solution was then purified by precipitation from petroleum ether 40/60 and remaining solvents were removed under vacuum. Mn(NMR)= 3200 g mol−1, Mw/Mn = 1.22. (Scheme 5.)
 | ||
| Scheme 5 General reaction conditions for reactive NHS-p(DEGMEMA) | ||
Bioconjugation
Before conjugation, hair samples were bleached to give a damaged surface. L'Oreal blue compact lightening powder (Platifiz Precision) (90 g) was mixed with oxidant crème (130 mL) until the solution became homogenous. This mixture was then applied to virgin hair samples and left to react for 30 min. The hair samples were then washed and the bleaching process repeated for a second time. Finally after washing to remove any remaining bleach, the samples were left to dry in air overnight.Reactive polymer (0.2 g, 4.22 × 10−5mol) was placed into a 100 mL round bottom flask with stirrer bar, and anhydrous DMSO (67.06 mL) and triethylamine (4.79 mL, 0.034 mol) was added. The solution was stirred for ten minutes before the damaged hair (0.44 g) samples were introduced. After 70 h stirring at ambient temperature, hair was collected and washed in deionised water at ambient temperature under gentle stirring for three days.
DSC sample preparation
Stainless steel sample pans were used for carrying out wet method DSC, 5 mg of hair was weighed into the pan and 50 μl of water was added. The pan was sealed shut with a washer and lid and then rotary mixed and left overnight for water to equilibrate throughout the sample. The samples were then held in the DSC at 100 °C for 3 min to remove thermal history and then heated to 180 °C at a scan rate of 5 °C/min. Aluminum sample pans were used for carrying out dry method DSC and 5 mg of hair was weighed into the pan and the pan was then sealed shut with a lid. The lid was subsequently pierced to allow moisture vapor escape during analysis. The samples were then heated in the DSC from 40 °C to 260 °C at a scan rate of 5 °C/min.Results and discussion
Polymer series 1: Bnz-p(PEGMEMA) and NHS-p(PEGMEMA)
The first series of control Bnz-p(PEGMEMA) (Scheme 2) and reactive NHS-p(PEGMEMA) (Scheme 3) polymers were synthesised with the mole ratio between initiator and monomer being the only parameter altered, to target a range of molecular weights, Table 1. It was of interest to synthesise a control polymer which would not react when placed in the presence of free amine residues present on hair. The benzyl group was introduced to the polymer by polymerisation from 2-bromo-2-methyl propionic acid benzyl ester initiator. In comparison, the reactive polymer is synthesised with an initiator containing an N-hydroxysuccinimdyl (NHS) ester. Polymerisations proceeded with first order kinetics, good control and gave polymers with narrow polydispersity index (PDI); but due to low initiator efficiency molecular weights were higher than predicted. This was taken into account when targeting different degrees of polymerisation (Fig. 2).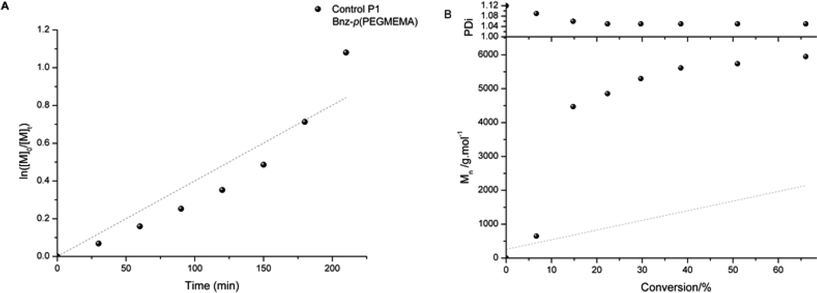 | ||
| Fig. 2 Kinetic plot of Control P1 Bnz-p(PEGMEMA) (a) and evolution of Mn and PDI with conversion (b) (grey points indicate theoretical Mn). Synthesis Conditions: CuBr, N-(n-ethyl)-2-pyridylmethanimine, toluene, 70 °C. | ||
| Polymer | Target DP | Mn (theory)/g mol−1 | Mn (NMR)/g mol−1 | Mn (GPC)a/g mol−1 | PDI | Conv./% |
|---|---|---|---|---|---|---|
| a Calibrated against PMMA standard. | ||||||
| Control P1 Bnz-p(PEGMEMA) | 6 | 2500 | 5000 | 7500 | 1.09 | 80 |
| Control P2 Bnz-p(PEGMEMA) | 10 | 3900 | 11900 |
14000 |
1.06 | 78 |
| Reactive P3 NHS-p(PEGMEMA) | 10 | 3400 | 10500 |
10300 |
1.09 | 67 |
| Reactive P4 NHS-p(PEGMEMA) | 20 | 9400 | 17800 |
13400 |
1.11 | 96 |
The control polymer P1 has benzylic end group protons observed at 7.37 ppm (Fig. 3). Reactive polymer P3 has NHS protons from the end group at 2.84 ppm (Fig. 4). When calculating the number of average molecular weight (Mn) by 1H NMR the initiator peak was integrated against the polymer peak. The peaks relating to the PEGMEMA monomer are quite broad at points ‘g’ and ‘f’ in the 1H NMR for both control and reactive polymers, due to the high number of PEG repeating units; however the CH2 group adjacent to the oxygen is very distinguishable and is used for calculation of the Mn. In addition, GPC was also used to measure molecular weight averages of the polymers.
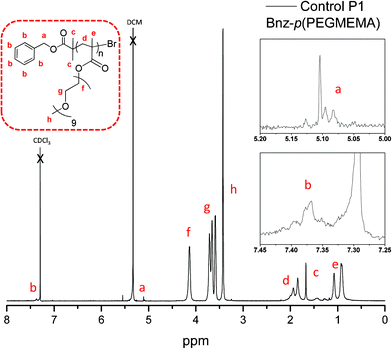 | ||
| Fig. 3 1H NMR of control P1: Bnz-p(PEGMEMA). | ||
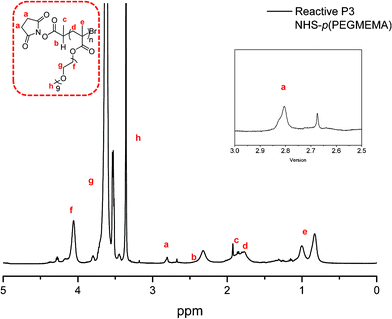 | ||
| Fig. 4 1H NMR of reactive P3: NHS-p(PEGMEMA). | ||
The distribution of polymer chains is seen in the MALDI-ToF spectrum with the ethylene glycol repeat unit with an equal spacing of 44 g mol−1 observed as lithium adducts (Fig. 5).
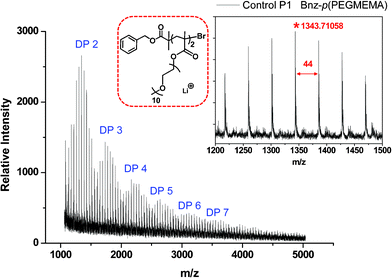 | ||
| Fig. 5 MALDI-ToF of control polymer P1: Bnz-p(PEGMEMA). | ||
Molecular weight averages were also measured using GPC and an acceptable correlation can be seen between the two methods (Table 1). The initiator used for the reactive polymers was selected due to its high reactivity; however, it has been shown previously to have lower initiator efficiency than a similar initiator which has a methyl group in place of the hydrogen atom labelled ‘b’ in Fig. 4.15
A comparison of the molecular weights and PDI of the polymers synthesised is shown in Fig. 6. P1 is the smallest polymer synthesised while P2 and P3 are higher molecular weight polymers. The trace for P4 reveals a slight shoulder towards higher molecular weight. This reaction was taken to a higher conversion and this has caused the PDI of the polymer to increase. This indicates termination from coupling reactions of polymer chains, as the concentration of polymer in solution is far greater than monomer at high conversion.
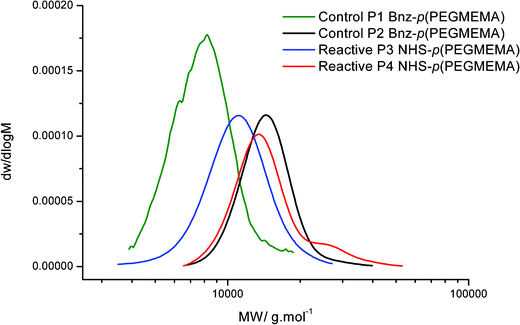 | ||
| Fig. 6 GPC traces showing the molecular weight distribution of PEGMEMA polymers. | ||
Polymer series 2: Bnz-p(DEGMEMA) and NHS-p(DEGMEMA)
For this series DEGMEMA was selected as it has a discrete structure, with two glycol repeating units, in comparison to PEGMEMA which has on average nine repeating units with a distribution of chain lengths. This structure therefore has a cleaner peak distribution in 1H NMR in comparison to the many repeating units of PEGMEMA. Control and reactive polymers were synthesised; Table 2.| Polymer | Target DP | Mn(theory)/g mol−1 | Mn(NMR)/g mol−1 | Mn(GPC)a/g mol−1 | PDi | Conv./% |
|---|---|---|---|---|---|---|
| a Calibrated against PMMA standards. | ||||||
| Control P5 Bnz-p(DEGMEMA) | 6 | 1600 | 4900 | 5300 | 1.13 | 56 |
| Control P6 Bnz-p(DEGMEMA) | 20 | 3800 | 5900 | 6900 | 1.21 | 95 |
| Control P7 Bnz-p(DEGMEMA) | 52 | 10000 |
10600 |
9100 | 1.14 | 57 |
| Reactive P8 NHS-p(DEGMEMA) | 6 | 1000 | 1500 | 2000 | 1.27 | 66 |
| Reactive P9 NHS-p(DEGMEMA) | 10 | 1000 | 1300 | 3300 | 1.23 | 45 |
| Reactive P10 NHS-p(DEGMEMA) | 20 | 1000 | 3200 | 4400 | 1.22 | 23 |
| Reactive P11 NHS-p(DEGMEMA) | 50 | 8300 | 25100 |
29600 |
1.28 | 85 |
| Reactive P12 NHS-p(DEGMEMA) | 100 | 15600 |
37500 |
46500 |
1.81 | 82 |
A variety of molecular weights were targeted and the molecular weights attained were higher in comparison to the theoretical values as expected. From the first polymer series the effect of reaction conversion on molecular weight and control was noticed. This was taken into account for series two and reactions were stopped prior to 90% conversion (with the exception of P6) to try and reduce this loss of control over Mn. The PDI for the reactive polymer P12 is much higher than the other polymers, indicating an increase in termination as molecular weight increases. The kinetic plot for control polymer P5 indicates first order kinetics followed, along with linear growth of Mn with conversion (Fig. 7).
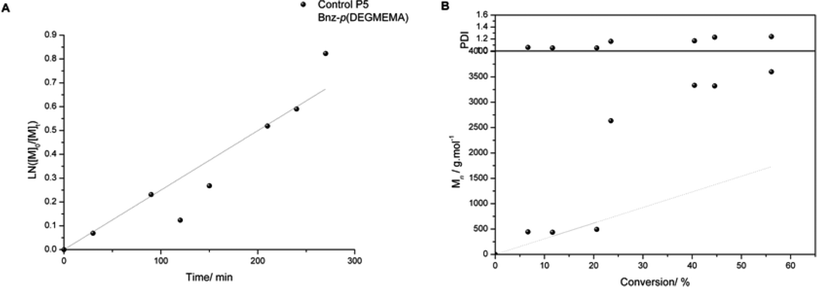 | ||
| Fig. 7 Kinetic plot of Control P5 Bnz-p(DEGMEMA) (a) and evolution of Mn and PDI with conversion (b). (Grey points indicate theoretical Mn). Synthesis Conditions: CuBr, N-(n-ethyl)-2-pyridylmethanimine, toluene, 70 °C. | ||
The 1H NMR of the polymer with the benzyl protons of the initiator fragment observed at 7.37 ppm (Fig. 8). The more discrete nature of the DEGMEMA monomer in comparison to PEGMEMA results in a clearer 1H NMR displaying each of the CH2 groups separately which are present in the ethylene glycol repeating units.
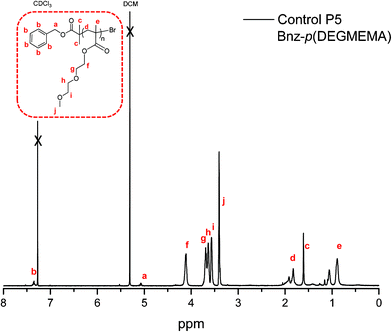 | ||
| Fig. 8 1H NMR of control P5: Bnz-p(DEGMEMA). | ||
The GPC traces for all polymers are unimodal except P11 and P12 where loss of control caused by reactions being taken to high conversion while targeting a high degree of polymerisation is observed (Fig. 9).
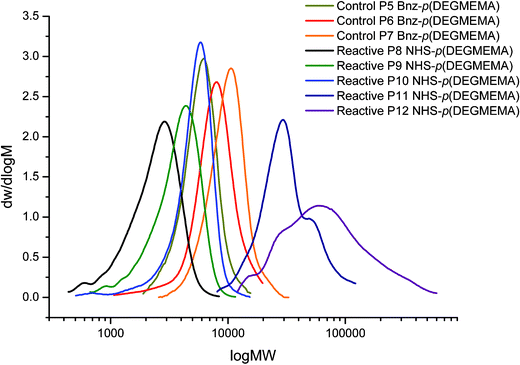 | ||
| Fig. 9 GPC traces showing MW distribution of DEGMEMA polymers. | ||
Bioconjugation
For polymer conjugation, α-keratin was selected as the biological surface. This protein is present in hair fibres and when hair becomes damaged by environmental factors such as bleaching or excess heat, the denaturation temperature of the protein decreases. Conjugation of a polymer to the protein can act as a barrier against such effects. Bioconjugation of the polymers to α-keratin was carried out via nucleophilic substitution (Scheme 6) with amine residues present in the protein, providing a hydrolytically stable amide link between the protein and the polymer.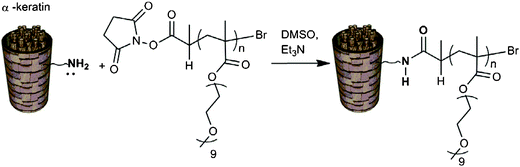 | ||
| Scheme 6 NHS–polymer conjugation to α-keratin. | ||
Polymers were reacted with damaged hair fibres in DMSO as solvent with triethylamine added and the reactions were left for 70 h to ensure efficient conjugation. DMSO was selected as solvent, rather than water so as to avoid any competing reaction during the nucleophilic substitution. Afterwards, hair was washed with distilled water (regularly changed) for three days to remove any unreacted polymer from the hair surface.
Differential scanning calorimetry (DSC) analysis
Unmodified hair was artificially damaged using controlled laboratory techniques to provide a damaged surface for conjugation and the denaturation temperature (Td) recorded via differential scanning calorimetry (DSC). After conjugation, the change in Td of the conjugate was recorded to compare against the Td of damaged hair (Fig. 10, Table 3). The Td is the point in which denatured helices, which were held in place by bonding, lose their structural integrity as these bonds are broken down and the complex protein matrix degrades.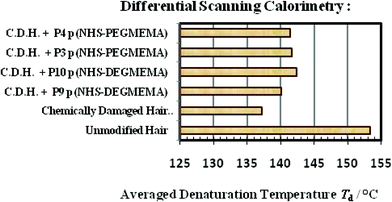
| Polymer | Hair Fibre | Averaged denaturation temperature/°C |
|---|---|---|
| — | Unmodified hair fibre | 153.4 |
| — | Chemically damaged hair fibre | 137.2 |
| Reactive P9 NHS-p(DEGMEMA) | Chemically damaged hair fibre | 140.1 |
| Reactive P10 NHS-p(DEGMEMA) | Chemically damaged hair fibre | 142.3 |
| Reactive P3 NHS-p(PEGMEMA) | Chemically damaged hair fibre | 141.6 |
| Reactive P4 NHS-p(PEGMEMA) | Damaged hair fibre | 141.4 |
Hair is hygroscopic, absorbing water from moisture in the air, which causes the protein structure to swell as the intermolecular hydrogen bonding between protein–protein bonds are broken and new hydrogen bonding between the protein and the water molecules are formed. Furthermore, hair strands damaged via bleaching results in the tertiary structure of the protein being modified. Bleaching disrupts the disulphide bonds in the protein, leading to an increase in concentration of cysteic acid and a decrease in protein matrix viscosity. The introduction of water breaks down any newly formed electrostatic interactions as well as hydrogen bonding and hence DSC analysis was carried out under aqueous conditions to mimic this environment.
DSC experiments demonstrate that damaged hair has a lower Td in comparison to unmodified hair. In all cases, α-keratin–polymer conjugates show an improved Td value for damaged hair. This indicates that the polymer successfully shields and protects the tertiary structure of the protein against environmental damage.
Scanning electron microscopy (SEM)
SEM was used to visualise the polymer coatings over the biological surface. Fig. 11 (A) shows the raised cuticle of the hair fibre caused by chemical damage. Image (B) shows the result of conjugation with the non-reactive control P1, with no observable polymer on the surface. Image (C) and (D) show conjugation with reactive P9 and P2 respectively. In both cases polymer is observed on the surface. Image (C) shows the polymer on the surface with the raised cuticle edge still exposed, while in image (D) the polymer is completely covering the raised cuticle. | ||
| Fig. 11 SEM images of chemically damaged hair reacted with polymers: (A) damaged hair surface, (B) damaged hair surface reacted with control P1, (C) damaged hair surface reacted with P9, (D) damaged hair surface reacted with P2. | ||
Conclusions
In summary, it has been shown that it is possible to target and synthesise control and reactive DEGMEMA and PEGMEMA polymers which conjugate to α-keratin protein. Through the use of living radical polymerisation it was possible to gain control over the polymer chain length and PDI. The conjugation reaction was facile with the conditions tuned for multi-site attachment to the protein as required in this application. Furthermore, it has been demonstrated that following multi-site conjugation, the thermal properties of damaged hair was improved, indicating sufficient polymer shielding. Finally, SEM provided visualization of the polymer coating on the protein surface.Acknowledgements
The authors thank EPSRC and Unilever PLC (SS) for funding. Equipment used in this research was partly obtained, through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).References
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS.
- Y. Kodera, A. Matsushima, M. Hiroto, H. Nishimura, A. Ishii, T. Ueno and Y. Inada, Prog. Polym. Sci., 1998, 23, 1233 CrossRef CAS.
- P. K. Working, M. S. Newman, J. Johnson and J. B. Cornacoff, Poly(Ethylene Glycol), 1997, 680, 45 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591 RSC.
- C. S. Fishburn, J. Pharm. Sci., 2008, 97 Search PubMed.
- A. Abuchowski, J. R. Mccoy, N. C. Palczuk, T. Vanes and F. F. Davis, J. Biol. Chem., 1977, 252, 3582 CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563 RSC.
- K. Velonia, Polym. Chem., 2010, 1, 944 RSC.
- J. P. Magnusson, A. O. Saeed, F. Fernandez-Trillo and C. Alexander, Polym. Chem., 2011, 2, 48 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004, 2026 Search PubMed.
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150 CrossRef CAS.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272 Search PubMed.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220 CrossRef CAS.
- C. T. Sayers, G. Mantovani, S. M. Ryan, R. K. Randev, O. Keiper, O. I. Leszczyszyn, C. Blindauer, D. J. Brayden and D. M. Haddleton, Soft Matter, 2009, 5, 3038 RSC.
- S. M. Ryan, X. X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697 Search PubMed.
- J. Nicolas, E. Khoshdel and D. M. Haddleton, Chem. Commun., 2007, 1722 Search PubMed.
- J. N. Cao and F. Leroy, Biopolymers, 2005, 77, 38 CrossRef CAS.
- V. F. Monteiro, A. P. Maciel and E. Longo, J. Therm. Anal. Calorim., 2005, 79, 289 CrossRef CAS.
- H. Zahn, J. Fohles, M. Nienhaus, A. Schwan and M. Spel, Ind. Eng. Chem. Prod. Res. Dev., 1980, 19, 496 CrossRef CAS.
- C. Popescu and H. Hocker, Chem. Soc. Rev., 2007, 36, 1282 RSC.
- A. D. Mclachlan, J. Mol. Biol., 1978, 124, 297 CrossRef CAS.
- V. M. Longo, V. F. Monteiro, A. S. Pinheiro, D. Terci, J.S. Vasconcelos, C. A. Paskocimas, E. R. Leite, E. Longo and J. A. Varela, Int. J. Cosmet. Sci., 2005, 28, 95 CrossRef.
- S. D. Bringans, J. E. Plowman, J. M. Dyer, S. Clerens, J. A. Vernon and W. G. Bryson, Exp. Dermatol., 2007, 16, 951 CrossRef CAS.
- L. Kreplak, A. Franbourg, F. Briki, F. Leroy, D. Dalle and J. Doucet, Biophys. J., 2002, 82, 2265 CrossRef CAS.
- F. J. Wortmann, C. Popescu and G. Sendelbach, Biopolymers, 2006, 83, 630 CrossRef CAS.
- F. J. Wortmann, M. Stapels, R. Elliott and L. Chandra, Biopolymers, 2006, 81, 371 CrossRef CAS.
- J. V. Gruber, B. R. Lamoureux, N. Joshi, L. Moral and M. A. Baak, Colloids Surf., B, 2000, 19, 127 CrossRef CAS.
- J. F. Calello, S. Kwan, A. Lukacs (III), A. A. Patil, B. A. Wolf and G. H. Armstrong, 2001, US 6277358.
- U. Bernecker, M. Ehlert and D. Hollenberg, 1999, DE 19752837.
- J. Chisem, E. Khoshdel, P. H. Neill, S. Pascual and E. S. Reid, 2002, WO 2002013773.
- J.-A. Allwohn, S. Birkel and A. Beyer, 1999, WO 9933434.
- Abstracts, P. Somasundaran, S. Chakraborty, Q. Qiang, P. Deo, J. Wang and R. Zhang, Int. J. Cosmetic. Sci., 2005, 27, 135 CrossRef.
- V. M. Longo, V. F. Monteiro, A. S. Pinheiro, D. Terci, J. S. Vasconcelos, C. A. Paskocimas, E. R. Leite, E. Longo and J. A. Varela, Int. J. Cosmet. Sci., 2006, 28, 95 CrossRef CAS.
- R. N. Keller and H. D. Wycoff, Inorg. Synth., 1946, 1 Search PubMed.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00157d |
| This journal is © The Royal Society of Chemistry 2011 |