Development of LPS antagonistic therapeutics: synthesis and evaluation of glucopyranoside-spacer-amino acid motifs†
Sophon
Kaeothip
,
Geeta
Paranjape
,
Shana E.
Terrill
,
Aileen F. G.
Bongat
,
Maria L. D.
Udan
,
Teerada
Kamkhachorn
,
Hope L.
Johnson
,
Michael R.
Nichols
* and
Alexei V.
Demchenko
*
Department of Chemistry and Biochemistry, University of Missouri – St. Louis, One University Boulevard, St. Louis, MO 63121, USA. E-mail: nicholsmic@umsl.edu; demchenkoa@umsl.edu
First published on 18th July 2011
Abstract
Sepsis is a serious medical condition characterized by bacterial infection and a subsequent massive systemic inflammatory response. The release of proinflammatory products and mediators from responding innate immune cells, such as mononuclear phagocytes, directly contributes to the pathogenesis of sepsis. The primary bacterial trigger of inflammation is lipopolysaccharide (LPS), which interacts with the germline-encoded macrophage receptor cluster of differentiation 14 (CD14) via its Lipid A moiety. In an effort to identify compounds that block LPS-induced inflammation we investigated a series of Lipid A analogs that lack a disaccharide core yet still possess potent antagonistic activity against LPS. We found it beneficial to develop molecules that contain the following: a glucopyranoside core, hydrophobic ether substituents, and an amino acid to provide an ionic character to the constructs. Here we report an efficient synthesis of molecules of this type and the ensuing biological studies thereof.
Introduction
Septicemia is a serious world-wide health problem associated with mortality rates of 40–60%.1 It has been estimated that 1% of hospital patients and 20–30% of ICU patients develop sepsis. The cardiovascular consequences of septic shock resulting from bacterial infections include myocardial dysfunction that develops in nearly all patients,2 vascular tone and permeability abnormalities, as well as abnormal oxygen delivery and metabolism. As a result, vital organs such as the brain, heart, kidneys, and liver may be affected or may fail, and this reflects in over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually in the US.1 It is well established that septic shock is initiated by the introduction of a bacterial endotoxin (or lipopolysaccharide, LPS) into the blood stream. LPS (Fig. 1), a vital component of the outer leaflet of the gram-negative outer membrane, has been shown to be a principle mediator of the depression of left ventricular function and myocardial contractility.3 LPS is comprised of three structural regions. One of these, the Lipid A region, consists of a polyacylated glucosamine disaccharide and is largely responsible for the toxic activity.4 The results of recent studies suggest that the ensuing proinflammatory response to LPS is by far more dangerous than the mere presence of LPS in circulation.5,6 LPS exerts its effects via interaction with a plasma LPS-binding protein (LBP), which has strong affinity for both the Lipid A region of the endotoxin and glycosylphosphatidyl inositol-anchored LPS receptor CD14 on mononuclear phagocytes. The LPS-LBP complex then interacts with CD14 followed by further complex formation with Toll-like receptor 4 (TLR4) and its co-receptor MD-2.7 TLR4 is an integral membrane protein that transmits the LPS signal to the inside of the cell and initiates the signaling pathways that lead to production of proinflammatory molecules, such as the cytokine, tumor necrosis factor α (TNFα).
000 deaths annually in the US.1 It is well established that septic shock is initiated by the introduction of a bacterial endotoxin (or lipopolysaccharide, LPS) into the blood stream. LPS (Fig. 1), a vital component of the outer leaflet of the gram-negative outer membrane, has been shown to be a principle mediator of the depression of left ventricular function and myocardial contractility.3 LPS is comprised of three structural regions. One of these, the Lipid A region, consists of a polyacylated glucosamine disaccharide and is largely responsible for the toxic activity.4 The results of recent studies suggest that the ensuing proinflammatory response to LPS is by far more dangerous than the mere presence of LPS in circulation.5,6 LPS exerts its effects via interaction with a plasma LPS-binding protein (LBP), which has strong affinity for both the Lipid A region of the endotoxin and glycosylphosphatidyl inositol-anchored LPS receptor CD14 on mononuclear phagocytes. The LPS-LBP complex then interacts with CD14 followed by further complex formation with Toll-like receptor 4 (TLR4) and its co-receptor MD-2.7 TLR4 is an integral membrane protein that transmits the LPS signal to the inside of the cell and initiates the signaling pathways that lead to production of proinflammatory molecules, such as the cytokine, tumor necrosis factor α (TNFα).
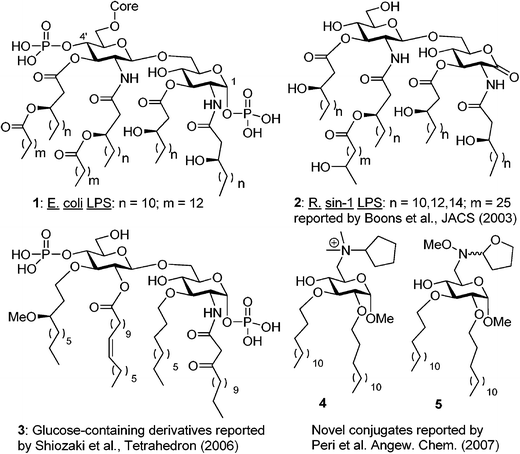 | ||
| Fig. 1 Structures of the Lipid A region of E. coli LPS (1 and structural mimetics thereof (2–5). | ||
Recent advances in the understanding of LPS structure-function relationships have provided some clues on the structural determinants responsible for the endotoxic activity of Lipid A.8 These determinants include the number and chain length of fatty acids (lipids), the disaccharide core, and the 1,4′-diphosphate groups of the E. coli type (1, Fig. 1). The fair stability (chemical or in vitro) of this class of compounds has been a major drawback in their synthesis and application. Although the exact role of the phosphate moieties is still unknown, the observation that a 1-hydroxyl-4′-O-phosphate derivative was inactive gave rise to a belief that the omission of at least one phosphate results in a complete loss of activity.8,9
In an effort to explore molecules that antagonize LPS signaling without activating the inflammatory cascade, our laboratory has designed simplified Lipid A analogs that lack the complexity of the highly lipidated diphosphorylated disaccharide core yet still maintain potent antagonistic activity against LPS. Herein we present the synthesis and unprecedented LPS-antagonistic activity of the methyl glucopyranoside-amino acid conjugates. This project was inspired by published reports of compounds that are structurally dissimilar to Lipid A yet still exhibit potent antagonistic activity. Amongst a myriad of research articles that have been disseminated in the past years, the following three noteworthy discoveries served as a justification and guide for our own studies.
First, as reported by Boons et al., a strong binding and antagonistic effect was achieved even with ‘phosphateless’ Rhizobium syn-1 disaccharide 2, which bears a lactone moiety at the reducing end (Fig. 1).10,11 This finding indicated that one could avoid hurdles associated with labile phosphate group installation, handling, and storage. Second, Shiozaki et al. showed that a strong anti-LPS antagonistic response can be achieved, even with disaccharide 3, in which the non-reducing glucosamine is replaced with glucose.12 In addition, instead of chiral β-hydroxy/acyl esters, relatively simple alkyl chains were used at the C-3 and 3′ positions, whereas a β-ketoamide moiety was used at C-2.
Third, Peri et al. demonstrated that even simple methyl glycoside-heterocycle conjugates 4 and 5 which bear alkyl chains instead of traditional lipids provide antagonistic activity.13 To this end, we found it attractive to develop molecules that contain the following: a methyl glucoside monosaccharide core to simplify the synthesis, hydrophobic chains to facilitate membrane intercalation, and an amino acid to provide an ionic character. Here we report an efficient synthesis of conjugates of this type and biological studies of their endotoxic activity in vitro.
Results and discussion
The synthesis began with the conversion of methyl α-D-glucopyranoside 6 into methyl 2,3-di-O-benzyl-α-D-glucopyranoside 714via sequential 4,6-benzylidene acetal formation, 2,3-dibenzylation, and acetal cleavage accomplished in 67% yield over three steps (Scheme 1). 4,6-Diol 7 was then regioselectively succinoylated at the primary position with succinic anhydride in the presence of 4-dimethylaminopyridine (DMAP) in pyridine to afford the derivative 8 in 71% yield. The carboxyl moiety of the linker was then coupled with 3-hydroxyl of the protected L-serine derivative 9.15,16 This was accomplished using N,N'-diisopropylcarbodiimide (DIC) as the coupling reagent in the presence of DMAP in pyridine to afford conjugate 10 in 80% yield.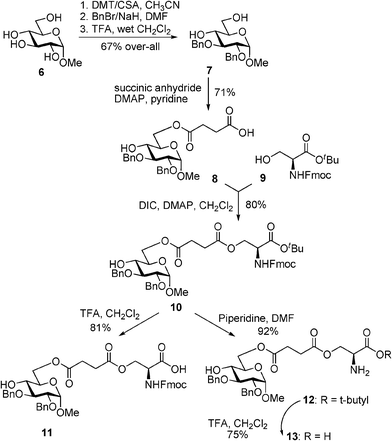 | ||
| Scheme 1 Synthesis of monosaccharide–aminoacid conjugates 10–13. | ||
Having obtained the key construct 10, we decided to pursue further functional group transformations. The main driving force for these synthetic manipulations was to obtain a series of simple analogs that would allow us to investigate the effect of cationic and anionic character on LPS-antagonistic activity in vitro. With this objective in mind, carboxylated compound 11 was obtained from 10 by cleavage of tert-butyl ester in the presence of TFA/DCM in 81% yield. Alternatively, the Fmoc protecting group could also be removed from compound 10 with piperidine in DMF to afford free amine 12. Subsequently, we also obtained compound 13 with both carboxyl and amine groups unprotected. This was accomplished by the treatment of compound 12 with TFA/CH2Cl2 to give derivative 13 in 75% yield.
The inhibitory activity of compounds 10–13 on LPS-induced TNFα production was investigated in vitro using THP-1 macrophages prepared as described in the Experimental section. THP-1 cells are an excellent system for studying inflammatory processes and serve as a model for peripheral monocytes/macrophages and their responses to bacterial infection. Compounds 10, 12, and 13 exhibited no inhibitory activity against LPS-induced TNFα production in the concentration range of 0.1 nM to 10 μM (Fig. 2A–C). These compounds were also tested in the absence of LPS and demonstrated no agonist activity (data not shown). Compound 11, which has a free carboxylic group, was able to significantly inhibit LPS-induced TNFα production at concentrations greater than 10 μM (Fig. 3). Unfortunately, cell viability measurements using an XTT reduction assay indicated that compound 11 was toxic to the cells in the 30–100 μM range. Although there was a notable gap between inhibition and toxicity at 30 and 50 μM, the similarities between the inhibition and toxicity curves suggested that much of the antagonistic activity by 11 was related to toxicity.
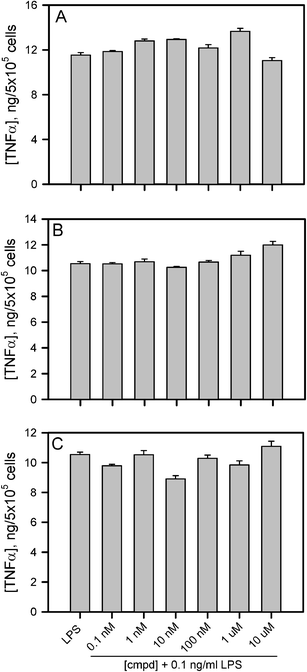 | ||
| Fig. 2 Compounds 10, 12, and 13 do not display LPS antagonistic activity. THP-1 human monocytes were treated as described in the Experimental section with 10 ng mL−1 PMA for 24 h at 37 °C and 5% CO2. The medium was then removed and adherent macrophages were further treated as described with increasing concentrations of compounds 10 (panel A), 12 (panel B) and 13 (panel C) for 30 min followed by incubation with 10 ng mL−1 LPS for 6 h. Secreted TNFα was measured by ELISA and the levels (ng mL−1) were reported on the y-axis after normalization by the number of counted macrophages (cells mL−1). Error bars represent the standard error (std err) for 3 different TNFα measurements from each cell treatment. | ||
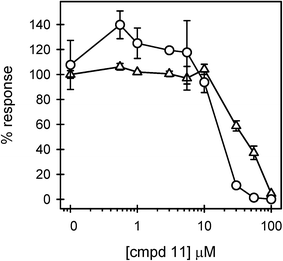 | ||
| Fig. 3 Compound 11 displays LPS antagonistic activity and cell toxicity. THP-1 macrophages were treated as described in the legend of Fig. 2, with increasing concentrations of compound 11. Secreted TNFα levels (circles) are the average ± std err for n = 2 trials and are reported as the % response of LPS in the absence of an antagonist compound. Immediately following the cell treatment described above and in the Fig. 2 legend, the macrophage viability (triangles) (n = 2 trials) was determined by XTT reduction as described in the Experimental. Cell viability is presented as a percentage of the cell viability measured in the absence of antagonists. | ||
With the purpose of gaining further insight into the effect of various substituents on the endotoxic activity of monosaccharide-amino acid conjugates, we obtained an analog of compound 11 in which benzyl groups have been replaced with acyl (myristoyl, C14) fatty acid chains. The synthesis of lipidated analog 19 was accomplished as depicted in Scheme 2. Methyl glycoside 6 was protected as 4,6-O-(p-methoxybenzylidene) acetal 14 by treatment with anisaldehyde dimethylacetal in the presence of camphorsulfonic acid in 89% yield. 14 was acylated with myristoyl chloride in the presence of the pyridine furnished compound 15 in 85% yield. The benzylidene ring in 15 was reductively opened by treatment with BH3-THF catalyzed with Cu(OTf)2 to obtain the unexpected product 16, which was lacking the C-2 acyl chain, in 67% yield. The loss of the acyl chain was rather unexpected, but this glitch was overcome by regioselective acylation with succinic anhydride in pyridine at the primary C-6 position followed by C-2 acylation with myristoyl chloride. This two-step one-pot procedure allowed us to obtain compound 17 in 79% yield.
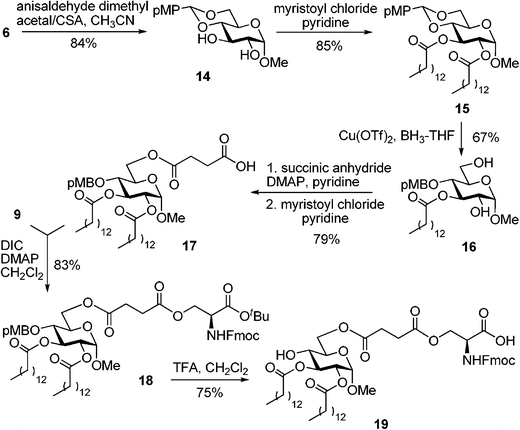 | ||
| Scheme 2 Synthesis of monosaccharide–fatty acid–amino acid conjugate 19, a lipidated analog of compound 11. | ||
L-serine derivative 915 was linked to the carboxyl group of 17viaDIC-mediated coupling in the presence of DMAP. The monosaccharide–fatty acid–amino acid conjugate 18 was obtained in 83% yield. Acid treatment of the fully protected compound 18 led to concomitant cleavage of the p-methoxybenzyl (pMB) group at C-4 and tert-butyl ester. As a result, compound 19 was isolated in 75% yield.
For comparative biological studies, we also accomplished the synthesis of the alkylated analog 24 as depicted in Scheme 3. The intermediate 14 was di-alkylated at C-2 and C-3 with myristyl bromide in the presence of NaH to afford compound 20 in 72% yield. The benzylidene ring in 20 was reductively opened by treatment with BH3-THF catalyzed with Cu(OTf)2 to obtain product 22 in 87%. Acylation of 22 with succinic anhydride led to compound 22 in 89% yield. L-serine derivative 915 was then linked to the carboxyl group of 22viaDIC-mediated coupling in the presence of DMAP. The resulting monosaccharide-amino acid conjugate 23 was obtained in 79% yield. Acid treatment of the fully protected compound 23 led to concomitant cleavage of the pMB group at C-4 and tert-butyl ester. As a result, target compound 24 was isolated in 81% yield.
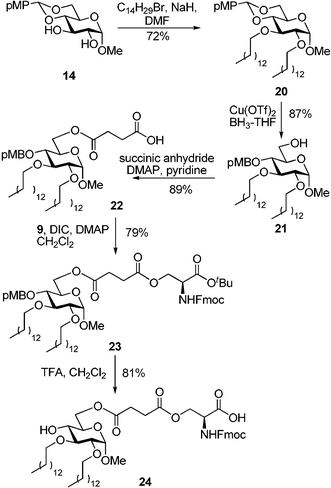 | ||
| Scheme 3 Synthesis of the monosaccharide–alkyl–amino acid conjugate 24, an alkylated analog of compound 11. | ||
Having obtained compounds 19 and 24, lipidated and alkylated analogs of compound 11, respectively, we were well positioned to perform comparative studies. In addition, as the comparison point, we obtained the standard positive control compound 4 as described by Peri et al.13 The lipidated compound 19 displayed a marked improvement in LPS-antagonistic ability. Inhibition of LPS-induced TNFα was found beginning at 550 nM 19 (Fig. 4). A reproducible biphasic response was consistently seen in the activity of 19. This may indicate multiple binding sites on the macrophages, both higher and lower affinity for the antagonist compound. 80% of the LPS response was blocked with 5 μM of compound 19 with no observable toxicity. Cell viability began to be compromised at 10 μM and some agonist activity was found in the 10–30 μM range (Fig. 4).
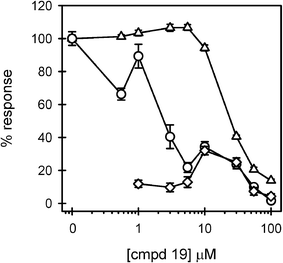 | ||
| Fig. 4 Compound 19 displays more potent LPS antagonistic activity. THP-1 macrophages were treated in 3 separate experiments as described in the Fig. 2 legend, with increasing concentrations of compound 19. Secreted TNFα levels were determined in the presence (circles) (n = 9 trials) and absence (diamonds) (n = 6 trials) of LPS. Cell viability (triangles) (n = 9 trials) was also assessed and presented as described in the Fig. 3 legend. | ||
The alkylated compound 24 was found to be a very effective inhibitor of LPS-induced TNFα production in human macrophages (Fig. 5). 70% inhibition was observed at 1 μM of 24 and overall inhibition reached 90% at 40 μM. Compound 24 exhibited no toxicity or agonist activity in the tested 0.2 to 40 μM range. Curve fitting of the inhibition data in Fig. 5 produced an IC50 value of 470 nM. These results were superior to those obtained with compound 413 which had an inhibition range from 3–10 μM and began to show agonist activity at concentrations > 10 μM (Fig. 6). Compound 4 was only toxic at high concentrations.
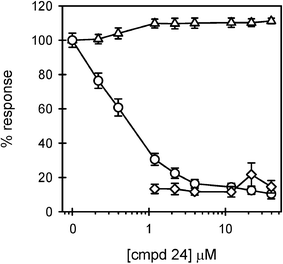 | ||
| Fig. 5 Compound 24 displays significant LPS antagonistic activity without toxicity. THP-1 macrophages were treated in 4 separate experiments, as described in the Fig. 2 legend, with increasing concentrations of compound 24. Secreted TNFα levels were determined in the presence (circles) (n = 12 trials) and absence (diamonds) (n = 8 trials) of LPS. Cell viability (triangles) (n = 12 trials) was also assessed and presented as described in the Fig. 3 legend. | ||
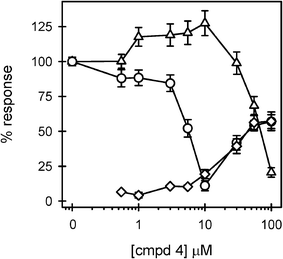 | ||
| Fig. 6 LPS antagonistic activity displayed by compound 4. THP-1 macrophages were treated in 3 separate experiments, as described in the Fig. 2 legend, with increasing concentrations of compound 4. Secreted TNFα levels were determined in the presence (circles) (n = 8 trials) and absence (diamonds) (n = 5 trials) of LPS. Cell viability (triangles) (n = 11 trials) was also assessed and presented as described in the Fig. 3 legend. | ||
The potent inhibition displayed by compound 24 of LPS-induced TNFα production without associated toxicity establishes this compound and those in its class as potential therapeutic and research compounds. These results extend the work of Boons and co-workers with phosphateless Lipid A derivatives11 by transitioning from disaccharide-based to monosaccharide-based compounds. The IC50 inhibition constant of 470 nM for compound 24 is the lowest reported for a monosaccharide compound and its potency in human macrophages significantly improves on the monosaccharide compounds tested in mouse bone-marrow-derived macrophages by Peri et al.13 Our monosaccharide-based compound in the current form is not as potent as some of the best disaccharide compounds such as E5531 (IC50 1.2 nM), E5564 (IC50 1.1 nM) and E5564 analogues (IC50 0.97 nM).8,12,17 This difference in potency is balanced by the ease of synthesis and modification of monosaccharide-based compounds. In fact, the observation that the replacement of the acyl chains in 19 with alkyl chains (24) produced a significantly more potent antagonist with less toxicity suggests that additional modifications may further enhance the activity. From these studies, it also became clear that subtle structural changes have an impact on whether a particular compound displays antagonist or agonist activity. The balance between these two activities has been explored before18 and suggests that small modifications to the compounds described in this investigation may enhance agonist activity, thus providing a basis for adjuvant development.19–21
In summary, studies described herein provide new information regarding the structure–activity relationship of a novel class of compounds that mimic classic Lipid A analogs, and the scientific basis for future studies to prevent the deleterious effects of endotoxemia. We believe that the results of these studies summarized in Table 1 will have far reaching impact on the treatment of patients diagnosed with endotoxemia. As a consequence, we can foresee additional efforts to investigate the molecular mechanisms underlying the antagonistic actions of the synthetic Lipid A analogs.
| Compound | TNFα inhibition (IC50) | Toxicity | Agonist activity |
|---|---|---|---|
| 10, 12, 13 | None observed | None observed | None observed |
| 11 | 46 μM | >10 μM | None observed |
| 19 | 3 μM | >10 μM | 10–30 μM |
| 24 | 0.5 μM | None observed | None observed |
| 4 | 6 μM | >30 μM | 30–100 μM |
Experimental
General
Column chromatography was performed on silica gel 60 (70–230 mesh) and reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at < 40 °C. CH2Cl2 and CH3CN were distilled from CaH2 directly prior to application. Pyridine was dried by refluxing with CaH2 and then distilled and stored over molecular sieves (3 Å). Cu(OTf)2 was co-evaporated with toluene (3 × 10 mL) and dried in vacuo for 2–3 h directly prior to application. N,N'-Diisopropylcarbodiimide, 4-dimethylaminopyridine, piperidine, and anhydrous DMF were used without further conditioning. Optical rotations were measured by a ‘Jasco P-1020’ polarimeter. Unless noted otherwise, 1H-NMR spectra were recorded in CDCl3 at 300 MHz (Bruker Avance) or at 500 MHz (Bruker ARX-500), 13C-NMR spectra and two-dimensional experiments were recorded in CDCl3 at 75 MHz (Bruker Avance) or at 125 MHz (Bruker ARX-500). HR FAB-MS determinations were made with the use of JEOL MStation (JMS-700) Mass Spectrometer, matrix m-nitrobenzyl alcohol, with NaI as necessary.Methyl 2,3-di-O-benzyl-6-O-(3-carboxypropanoyl)-α-D-glycopyranoside (8)
4-Dimethylaminopyridine (DMAP, 32 mg, 0.26 mmol) and succinic anhydride (0.16 g, 1.56 mmol) were added to a stirred solution of methyl 2,3-di-O-benzyl-α-D-glucopyranoside 714 (0.5 g, 1.3 mmol) in dry pyridine (5 mL) and the resulting mixture was stirred for 16 h at room temperature (rt) under argon. After that, the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CH2Cl2 (30 mL) and washed with water (3 × 15 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to give the title compound 8 (0.45 g, 0.95 mmol) as a colorless syrup in 71% yield. Analytical data for 8: Rf 0.43 (ethyl acetate); [α]22D +7.19o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 2.60 (m, 4H, 2 × CH2), 3.38 (s, 3H, OCH3), 3.44 (dd, 1H, J2,3 = 9.5 Hz, H-2), 3,51 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.73 (m, 1H, J5,6b = 4.8 Hz, H-5), 3.80 (dd, 1H, J2,3 = 9.2 Hz, H-3), 4.26 (dd, 1H, J6a,6b = 10.0 Hz, H-6a), 4.42 (dd, 1H, H-6b), 4.61 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.65 (d, 1H, 2J= 12.1 Hz, ½ CH2Ph), 4.74–4.79 (m, 2H, CH2Ph), 4.99 (d, 1H, 2J = 11.3 Hz, ½ CH2Ph), 7.26–7.36 (m, 10H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 55.4, 63.7, 69.4, 69.9, 73.3, 75.3, 79.7, 81.0, 98.3, 128.0 (×2), 128.1 (×2), 128.2 (×4), 128.6 (×2), 128.7 (×2), 138.1, 138.6, 172.6, 177.2 ppm; HR FAB MS [M+Na]+ calcd for C25H30O9Na 497.1788, found 497.1770.Methyl 2,3-di-O-benzyl-6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-3-(tert-butoxy)-3-oxopropyl)oxy-4-oxobutanoyl)-α-D-glycopyranoside (10)
DMAP (12.7 mg, 0.01 mmol) and N,N'-diisopropylcarbodiimide (DIC, 0.16 mL, 1.04 mmol) were added to a stirred solution of O-tert-butyl N-fluorenylmethoxycarbonyl-L-serine ester 915,16 (0.19 g, 0.52 mmol) in CH2Cl2 (4 mL). After 1 h, carboxylic acid derivative 8 (0.29 g, 0.62 mmol) was added and the reaction was stirred for 5–9 h at rt until no further conversion of the starting material could be detected by TLC. The reaction mixture was diluted with CH2Cl2 (20 mL), washed with water (2 × 15 mL) and brine (15 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound 10 (0.34g, 0.41 mmol) as a white powder in 80% yield. Analytical data for 10: Rf 0.34 (ethyl acetate/hexane, 1/1, v/v); [α]27D +12.1o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.48 (s, 9H, t-Bu), 2.53 (d, 1H, J = 2.8 Hz, OH), 2.66 (m, 4H, 2 × CH2), 3.37 (s, 3H, OCH3), 3.42 (d, 1H, J2,3 = 9.0, H-2), 3.73 (m, 1H, H-5), 3.78 (dd, 2H, J3,4 = 9.1 Hz, H-3, CHFmoc), 4.25 (m, 2H, H-6b, SerCβ-H), 4.38 (m, 2H, H-6a, SerCβ-H), 4.44 (m, 2H, OCH2Fmoc), 4.52 (m, 1H, SerCα-H), 4.61 (d, 1H, J1,2 = 3.2 Hz, H-1), 4.64 (d, 1H, 2J = 12.1 Hz, ½ CH2Ph), 4.75 (dd, 2H, J = 12.0 Hz, CH2Ph), 4.98 (d, 1H, 2J = 11.3 Hz, ½ CH2Ph), 4.75 (d, 1H, JNH,CH = 8.0 Hz, Ser-NH), 7.27–7.77 (m, 18H, aromatic) ppm; 13C NMR (125 MHz, CDCl3): δ, 28.1 (×3), 29.1 (×2), 47.3, 54.1, 55.5, 63.9, 64.8, 67.4, 69.4, 70.1, 73.4, 75.7, 79.7, 81.3, 83.3, 98.4, 120.2 (×2), 125.4 (×2), 127.3, 127.9 (×2), 128.1, 128.2 (×2), 128.3 (×3), 128.7, 128.8 (×3), 138.2, 138.9 , 141.5 (×2), 144.0 (×2), 144.1, 156.0, 168.5, 171.8, 172.6 ppm; HR FAB MS [M+Na]+ calcd for C47H53NO13Na 862.3415, found 862.3432.Methyl 2,3-di-O-benzyl-6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-2-carboxyethyl)oxy-4-oxobutanoyl)-α-D-glycopyranoside (11)
Conjugate 10 (0.25g, 0.29 mmol) was dissolved in TFA/wet CH2Cl2 (1/5, v/v, 2 mL) and the resulting mixture was stirred for 2 h at rt. After that, the volatiles were evaporated under reduced pressure, the residue was diluted with CH2Cl2 (5 mL) and neutralized with triethylamine (until pH ~ 7). The volatiles were removed under reduced pressure and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane, 1/1, v/v) to afford the title compound 11 (0.18 g, 0.24 mmol) as a white foam in 81% yield. Analytical data for 11: Rf 0.5 (methanol/ethyl acetate, 1/4, v/v); [α]26D +22.5o (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3/(CD3)2SO, 2/1, v/v): δ = 2.49–2.54 (m, 4H), 3.26 (s, 3H, OCH3), 3.39 (m, 2H), 3.57 (m, 2H), 4.01 (br. s, 1H), 4.12 (m, 2H), 4.22 (m, 2H), 4.32 (dd, 2H, J = 6.6 Hz), 4.43 (d, 1H, J = 5.1 Hz), 4.60 (s, 2H), 4.76 (m, 2H), 7.24–7.93 (m, 18H, aromatic) ppm; 13C NMR (75 MHz, CDCl3/(CD3)2SO, 2/1, v/v): δ, 28.8, 28.9, 31.1, 47.0, 54.7, 54.8, 63.4, 63.9, 65.3, 65.9, 69.8, 70.2, 71.8, 74.5, 79.4, 81.5, 97.3, 120.5 (×2), 125.5 (×2), 127.4 (×2), 127.5, 127.8 (×4) , 127.9 (×4), 128.0 (×3), 128.3 (×3), 128.5 (×3), 138.9, 139.6, 141.1 (×2), 144.2, 156.0, 172.1, 172.2 (×2) ppm; HR FAB MS [M+Na]+ calcd for C43H45NO13Na 806.2789, found 806.2780.Methyl 2,3-di-O-benzyl-6-O-(4-((S)-2-amino-3-(tert-butoxy)-3-oxopropyl)oxy-4-oxobutanoyl)-α-D-glycopyranoside (12)
Piperidine (0.5 mL) was added dropwise to a solution of monosaccharide 10 (0.1 g, 0.12 mmol) in DMF (2.0 mL) and the resulting mixture was stirred for 20 min at rt. After that, the reaction mixture was concentrated in vacuo and co-evaporated with toluene (×3). The residue was purified by flash column chromatography (ethyl acetate/hexane, 1/1, v/v) to give the title compound 12 (67 mg, 0.1 mmol) as a yellow syrup in 92% yield. Analytical data for 12: Rf 0.62 (methanol/ethyl acetate, 1/9, v/v); [α]26D +17.3o (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 1.49 (s, 9H, t-Bu), 2.52–2.66 (m, 3H), 2.73–2.78 (m, 1H), 3.02 (broad s, 1H, OH), 3.38 (s, 3H, OCH3), 3.46 (d, 1H, J2,3 = 9.4 Hz, H-2), 3.50 (ddd, 1H, J4,5 = 9.5 Hz, H-4), 3.75 (m, 2H, H-5, Ser-CβH), 3.80 (m, 2H, H-3, Ser-CβH), 4.18 (dd, 1H, J6a,6b = 10.19 Hz, H-6a), 4.46 (m, 1H, Ser-CαH), 4.52 (dd, 1H, J5,6b = 4.19 Hz, H-6b), 4.61 (d, 1H, J1,2 = 3.53 Hz, H-1), 4.65 (d, 1H, 2J = 12.5 Hz, ½ CH2Ph), 4.77 (dd, 2H, J = 11.5 Hz, CH2Ph), 4.99 (d, 1H, 2J = 9.0 Hz, ½ CH2Ph), 6.49 (d, 1H, JNH,CH = 6.95 Hz, Ser-NH), 7.27–7.31 (m, 10H, aromatic) ppm; 13C NMR (125 MHz, CDCl3): δ, 28.2 (×3), 29.8, 31.0, 31.1, 55.5 (×2), 55.7, 63.3, 63.6, 69.5, 69.8, 73.5, 75.9, 79.8, 81.5, 83.0, 98.5, 128.1, 128.2, 128.3(×2), 128.4 (×2), 128.7 (×2), 138.2, 138.7, 169.6, 172.0, 173.1 ppm; HR FAB MS [M+Na]+ calcd for C32H43NO11Na 640.2734, found 640.2741.Methyl 2,3-di-O-benzyl-6-O-(4-((S)-2-amino-2-carboxyethyl)oxy-4-oxobutanoyl)-α-D-glycopyranoside (13)
Conjugate 12 (0.1 g, 0.12 mmol) was dissolved in TFA/wet CH2Cl2 (1/5, v/v, 2 mL) and stirred for 2 h. Upon completion as assessed by TLC analysis, solvents were evaporated in vacuo. The residue was dissolved in CH2Cl2 (5 mL) and neutralized with triethylamine (until pH ∼ 7). Solvents were removed under reduced pressure. The residue was subjected to column chromatography on silica gel (methanol–ethyl acetate gradient elution) to obtain the product 13 (50 mg, 0.09 mmol) as white foam in 75% yield. Analytical data for 13: Rf 0.5 (methanol/ethyl acetate, 1/4, v/v); 1H NMR (500 MHz, D2O): δ, 1.68 (m, 2H), 1.82 (m, 3H), 2.73 (m, 4H), 3.19 (dd, 3H, J = 5.7 Hz), 3.4 (s, 3H, OCH3), 3.58 (ddd, 2H, J = 9.5, 10.0 Hz), 3.80 (dd, 1H, J = 9.2 Hz), 3.87 (d, 3H, J = 4.7 Hz), 4.33–4.37 (m, 2H, J = 4.7 Hz), 4.54 (d, 1H, J = 10.9 Hz), 7.44 (s, 10H, aromatic) ppm; 13C NMR (75 MHz, D2O): δ 29.5, 30.5, 55.4 (×2), 61.4, 63.7, 69.7, 69.8 (×2), 73.4, 75.5, 79.1 (×2), 81.1, 97.8, 128.7, 128.8 (×3), 129.0, 129.1 (×3), 137.7, 137.9, 174.5 (×2), 174.6 ppm; HR FAB MS [M+Na]+ calcd for C28H35NO11Na 584.2108, found 584.2104.Methyl 4,6-O-(p-methoxybenzylidene)-2,3-di-O-tetradecanoyl-α-D-glucopyranoside (15)
Myristoyl chloride (2.34 mL, 8.67 mmol) and DMAP (70 mg, 0.57 mmol) were added to a stirred solution of methyl 4,6-O-(p-methoxybenzylidene)-α-D-glucopyranoside 1422 (1.0 g, 2.89 mmol) in pyridine (15 mL) at 0 °C. The mixture was stirred under an atmosphere of argon for 16 h. Upon completion, the reaction mixture was concentrated in vacuo. The residue was dissolved in CH2Cl2 (30 mL), and washed with 1N HCl (2 × 15 mL), water (2 × 15 mL) and brine (15 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford derivative 15 (1.81 g, 2.48 mmol) as a white foam in 86% yield. Analytical data for 15: Rf 0.47 (ethyl acetate/hexane, 3/7, v/v); [α]23D +23.6o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.88 (t, 6H, 2 × CH3), 1,26 (br. s, 40H, 20 × CH2), 1.59 (m, 4H, 2 × CH2), 2.29 (m, 4H, 2 × CH2), 3.40 (s, 3H, OCH3), 3.62 (dd, 1H, J4,5 = 9.7 Hz, H-4), 3.75 (dd, 1H, J6a,6b = 10.2 Hz, H-6a), 3.78 (s, 3H, OCH3), 3.92 (m, 1H, H-5), 4.28 (dd, 1H, J5,6b = 4.7 Hz, H-6b), 4.82 – 2.94 (m, 2H, H-1, 2), 5.46 (s, 1H, > CHPh), 5.60 (dd, 1H, J3,4 = 9.7 Hz, H-3), 6.86 (d, 2H, J = 8.8 Hz, aromatic), 7.25 (d, 2H, J = 8.7 Hz, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 14.3 (×2), 22.9 (×2), 25.1 (×2), 25.28, 29.2 (×2), 29.4, 29.5 (×3), 29.6, 29.7, 29.8 (×2), 29.9, 32.1 (×2), 34.3, 34.5, 55.4, 55.5, 62.5, 68.8, 68.9, 71.6, 76.8, 77.2, 77.6, 79.5, 97.8, 101.6, 113.7 (×2), 127.6 (×2), 129.62, 160.2, 172.6, 173.4ppm; HR FAB MS [M+Na]+ calcd for C43H72O9Na 755.5074, found 755.5090.Methyl 4-O-p-methoxybenzyl-3-O-tetradecanoyl-α-D-glucopyranoside (16)
A 1 M solution of BH3-THF in tetrahydrofuran (5 mL, 5 mmol) was mixed with compound 15 (0.75 g, 1.02 mmol) and the resulting mixture was stirred for 10 min at rt under argon. Freshly conditioned copper(II) trifluoromethanesulfonate (18 mg, 0.05 mmol) was added, and the reaction mixture was stirred for 2 h at rt. After that, the reaction mixture was cooled to 0 °C and then quenched by the sequential addition of triethylamine (0.14 mL, 1 mmol) and methanol (1.8 mL). The resulting mixture was concentrated under reduced pressure followed by co-evaporation with methanol. The residue was purified by flash column chromatography on silica gel (ethyl acetate–hexane gradient elution) to give the title compound 16 (0.35g, 0.67 mmol) as white solid in 67% yield. Analytical data for 16: Rf 0.40 (ethyl acetate/hexane, 7/3, v/v); [α]22D+78.5o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 0.88 (t, 3H, J = 7.02 Hz, CH3), 1.24 (br. s, 20H, 10 × CH2), 1.63 (m, 2H, CH2), 1.97 (br. s, 1H, OH), 2.23–3.82 (m, 3H, CH2, OH), 3.41 (s, 3H, OCH3), 3.52–3.82 (m, 5H, H-2, 4, 5, 6a, 6b), 3.79 (s, 3H, OCH3), 4.57 (dd, 2H, J = 12.4 Hz, CH2Ph), 4.75 (d, 1H, J1,2 = 3.7 Hz, H-1), 5.30 (dd, 1H, J = 9.1 Hz, H-3), 6.85 (d, 2H, J = 8.6 Hz, aromatic), 7.20 (d, 2H, J = 8.6 Hz, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ = 14.3, 22.9, 25.1, 29.3, 29.5 (×2), 29.6, 29.8 (×4), 29.9, 32.1, 34.7, 55.4, 55.5, 61.7, 71.0, 71.9, 74.3, 74.9, 99.6, 114.0 (×2), 129.6 (×2), 130.1, 159.5, 174.3 ppm; HR FAB MS [M+Na]+ calcd for C29H48O8Na 547.3247, found 547.3254.Methyl 6-O-(3-carboxypropanoyl)-4-O-p-methoxybenzyl-2,3-di-O-tetradecanoyl-α-D-glucopyranoside (17)
DMAP (8.1 mg, 0.06 mmol) and succinic anhydride (67 mg, 0.67 mmol) were added to a stirred solution of derivative 16 (0.35g, 0.67 mmol) in dry pyridine (5 mL) and the resulting mixture was stirred for 16 h at rt under argon. After that, myristoyl chloride (0.21 ml, 0.80 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at rt. After that, the resulting mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (30 mL) and washed successively with 1N HCl (2 × 15 mL), water (2 × 15 mL) and brine (15 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to give compound 17 (0.43 g, 0.52 mmol) as white syrup in 79% yield. Analytical data for 17: Rf 0.55 (ethyl acetate-hexane 1/1, v/v); [α]24D +42.4 o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.87 (t, 6H, 2 × CH3), 1.25 (br. s, 40H, 20 × CH2), 1.59 (m, 4H, 2 × CH2), 2.26 (m, 2H, CH2), 2.33 (m, 2H, CH2), 2.61 (m, 4H, 2 × CH2), 3.36 (s, 3H, OCH3), 3.59 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.90 (m, 1H, H-5), 4.28 (dd, 2H, J5,6b = 4.4 Hz, J6a,6b = 11.5 Hz, H-6a, 6b), 4.44 (d, 1H, 2J = 10.7 Hz, ½ CH2Ph), 4.54 (d, 1H, 2J = 10.7, ½ CH2Ph), 4.87 (m, 2H, H-1, 2), 5.55 (dd, 1H, J3,4 = 9.5 Hz, H-3), 6.84 (d, 2H, J = 8.7 Hz, aromatic), 7.16 (d, 2H, J = 8.6 Hz, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 14.3 (×3), 22.8 (×3), 25.1 (×2), 28.8, 29.2, 29.3, 29.4 (×2), 29.5, 29.6, 29.7 (×4), 29.8 (×4), 32.1 (×4), 34.2, 34.5, 55.4 (×2), 62.7, 68.6, 71.8, 71.9, 74.3, 75.7, 96.9, 114.0 (×2), 129.5, 129.8 (×2), 159.6, 171.7, 172.8, 173.7 ppm; HR FAB MS [M+Na]+ calcd for C47H78O12Na 857.5391, found 857.5410.Methyl 6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-3-(tert-butoxy)-3-oxopropyl)oxy-4-oxobutanoyl)-2,3-di-O-tetradecanoyl-α-D-glucopyranoside (18)
DMAP (9.5 mg, 0.07 mmol) and DIC (0.12 mL, 0.78 mmol) were added to a stirred solution of O-tert-butyl-N-fluorenylmethoxycarbonyl-L-serine 915,16 (0.15 g, 0.39 mmol) in CH2Cl2 (3 mL). After 1 h, the carboxylic acid derivative 17 (0.39 g, 0.46 mmol) was added and the reaction was stirred for 5–9 h at rt until no further conversion of the starting material could be detected by TLC. The reaction mixture was diluted with CH2Cl2 (20 mL), washed with water (2 × 10 mL) and brine (10 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford the title compound 18 (0.38 g, 0.32 mmol) as a white solid in 83% yield. Analytical data for 18: Rf 0.5 (ethyl acetate/hexane, 3/7, v/v); [α]24D +25.7o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.88 (t, 6H, 2 × CH3), 1.25 (br. s, 40H, 20 × CH2), 1.49 (s, 9H, t-Bu), 1.59 (m, 4H, 2 × CH2), 1.59 (m, 4H, 2 × CH2), 2.30 (m, 4H, 2 × CH2), 3.37 (s, 3H, OCH3), 3.48 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.76 (s, 3H, OCH3), 3.90 (m, 1H, H-5), 4.16–4.26 (m, 3H, H-6a, 6b, Ser-CβH), 4.35–4.55 (m, 6H, CH2Ph, Ser-CαH, CHFmoc, OCH2Fmoc), 4.90 (m, 2H, H-1, 2), 5.58 (dd, 1H, J3,4 = 9.7 Hz, H-3), 5.82 (d, 1H, JNH,CH = 8.3 Hz, Ser-NH), 6.18–7.77 (m, 12H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 14.2 (×3), 22.8 (×3), 25.0, 28.0 (×4), 29.3 (×2), 29.4 (×2), 29.5 (×3), 29.6 (×3), 29.8 (×5), 32.0 (×3), 34.2, 34.4, 47.2, 54.0, 55.3 (×2), 62.6, 67.4, 68.5, 71.7, 71.9, 74.3, 76.7, 83.0, 96.8, 113.9 (×3), 120.1 (×2), 125.4, 127.2 (×2), 127.8 (×2), 129.3 (×2), 129.6 (×2), 141.4 (×2), 144.0 (×2), 156.0, 159.5, 168.4, 171.5, 171.8, 172.7, 173.5 ppm; HR FAB MS [M+Na]+ calcd for C69H101NO16Na 1222.7018, found 1222.7023.Methyl 6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-2-carboxyethyl)oxy-4-oxobutanoyl)-2,3-di-O-tetradecanoyl-α-D-glucopyranoside (19)
Compound 18 (0.2 g, 0.16 mmol) was dissolved in TFA/wet CH2Cl2 (1/5, v/v, 4 mL) and stirred for 2 h at rt. After that, the volatiles were evaporated in vacuo, and the residue was diluted with CH2Cl2 (5 mL) and neutralized with triethylamine (until pH ~ 7). The volatiles were removed under reduced pressure and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane, 1/1, v/v) to afford the title compound 19 (0.13 g, 0.12 mmol) as a white foam in 75% yield. Analytical data for 19: Rf 0.5 (methanol-ethyl acetate 1/9, v/v); 1H NMR (300 Hz, CDCl3): δ, 0.88 (t, 6H, 2 × CH3), 1.25 (br. s, 40H, 20 × CH2), 1.58 (m, 4H, 2 × CH2), 2.33 (m, 4H, 2 × CH2), 2.64 (br. s, 4H, 2 × CH2), 3.27 (br. s, 1H, OH), 3.38 (s, 3H, OCH3), 3.47 (dd, 1H, J4,5 = 9.4 Hz, H-4), 3.83 (m, 1H, H-5), 4.21–4.51 (m, 7H, H-6a, 6b, Ser-CβH2, CHFmoc, OCH2Fmoc), 4.67 (m, 1H, Ser-CαH) 4.86 (m, 1H, H-2), 4.90 (br. s, 1H, H-1), 5.31 (dd, 1H, J3,4 = 9.1 Hz, H-3), 5.96 (br. s, 1H, JNH,CH = 8.3 Hz, Ser-NH), 7.27–7.76 (m, 8H, aromatic) ppm; 13C NMR (75 MHz, CDCl3); δ, 14.3 (×3), 22.9 (×3), 25.0 (×2), 25.1, 28.9, 29.2, 29.30, 29.4 (×2), 29.5 (×2), 29.6, 29.8 (×2), 29.9, 32.1 (×2), 34.2 (×2), 34.5, 47.2, 55.5, 62.9, 64.3, 67.6, 69.4, 69.6, 69.9, 71.2, 72.7, 77.4, 96.8, 114.1, 120.2 (×2), 124.9 (×2), 125.3 (×2), 127.3 (×2), 127.9 (×2), 141.4 (×2), 143.8, 143.9, 171.7, 171.8, 174.3, 174.6, 175.1 ppm; HR FAB MS [M+Na]+ calcd for C57H85NO15Na 1046.5817, found 1045.5822.Methyl 6-O-(3-carboxypropanoyl)-4-O-p-methoxybenzyl-2,3-di-O-tetradecanyl-α-D-glucopyranoside (22)
DMAP (7.0 mg, 0.05 mmol) and succinic anhydride (71 mg, 0.70 mmol) were added to a stirred solution of methyl 4-O-p-methoxybenzyl-2,3-di-O-tetradecanyl-α-D-glucopyranoside 2113 (0.40 g, 0.56 mmol) in dry pyridine (5 mL) and the resulting reaction mixture was stirred for 16 h at rt under argon. After that, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (30 mL) and washed successively with 1N HCl (2 × 15 mL), water (2 × 15 mL) and brine (15 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to give the title compound 22 (0.41 g, 0.50 mmol) as a colorless syrup in 89% yield. Analytical data for 22: Rf 0.55 (ethyl acetate/hexane, 7/3, v/v); [α]24D +42.9o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.88 (t, 6H, 2 × CH3), 1.26 (br. s, 40H, 22 × CH2), 1.63 (m, 4H, 2 × CH2), 2.64 (m, 2H, 2 × CH2), 3.30 (dd, 1H, J2,3 = 9.7 Hz, H-2), 3.38 (s, 3H, OCH3), 3.39 (m, H-4), 3.59–3.76 (m, 5H, H-3, 5, OCH2, OCH2a), 3.80 (s, 3H, OCH3), 3.86 (m, 1H, OCH2b), 4.28 (m, 2H, J5,6b = 4.7 Hz, J5,6a = 2.3 Hz, J6a,6b = 12.0 Hz, H-6a, 6b), 4.49 (d, 1H, 2J = 10.5 Hz, ½ CH2Ph), 4.76 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.81 (d, 1H, 2J = 10.5 Hz, ½ CH2Ph), 6.89 (d, 2H, J = 8.9 Hz, aromatic), 7.22 (d, 2H, J = 9.7 Hz, aromatic) ppm; 13C NMR (125 MHz, CDCl3): δ = 14.3 (×3), 22.9 (×3), 26.2, 26.5, 28.8, 28.9, 29.5 (×5), 29.7 (×4), 29.8, 29.9, 30.3, 30.8, 32.1 (×4), 55.3, 55.5, 63.6, 68.8, 72.0, 74.1, 74.8, 80.9, 81.9, 98.1, 114.1 (×3), 130.1 (×3), 130.4, 159.6, 172.0, 176.5 ppm; HR FAB MS [M+Na]+ calcd for C47H82O10Na 829.5805, found 829.5806.Methyl 6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-3-(tert-butoxy)-3-oxopropyl)oxy-4-oxobutanoyl)-2,3-di-O-tetradecanyl-α-D-glucopyranoside (23)
DMAP (6.2 mg, 0.05 mmol) and DIC (0.16 mL, 1.01 mmol) were added to a stirred solution of O-tert-butyl-N-fluorenylmethoxycarbonyl-L-serine 915,16 (0.39 g, 0.60 mmol) in CH2Cl2 (3 ml). After 1 h, the carboxylic acid derivative 22 (0.41 g, 0.50 mmol) was added and the reaction was stirred for 5–9 h until no further conversion of the starting material could be detected by TLC. The reaction mixture was diluted with CH2Cl2 (20 mL), washed with water (2 × 10 mL) and brine (10 mL). The organic phase was separated, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound 23 (0.47 g, 0.40 mmol) as a colorless amorphous solid in 79% yield. Analytical data for 23: Rf 0.43 (ethyl acetate/hexane, 3/7, v/v); [α]24D +36.1o (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.88 (t, 6H, 2 × CH3), 1.17 (m, 40H, 20 × CH2), 1.38 (s, 9H, t-Bu), 1.54 (m, 4H, 2 × CH2), 2.53 (m, 4H, 2 × CH2), 3.18 (dd, 1H, J2,3 = 9.7 Hz, H-2), 3.26 (s, 3H, OCH3), 3.28 (m, 1H, H-4), 3.42–3.66 (m, 5H, H-3, 5, OCH2, OCH2a), 3.67 (s, 3H, OCH3), 3.79 (m, 1H, OCH2b), 4.11–4.46 (m, 9H, H-6a, 6b, ½ CH2Ph, Ser-CαH, Ser-CβH2, , CHFmoc, OCH2Fmoc), 4.66 (d, 1H, J1,2 = 3.3 Hz, H-1), 4.71 (d, 1H, 2J = 10.5 Hz, ½ CH2Ph), 5.71 (d, 1H, JNH,CH = 8.1 Hz, Ser-NH), 6.75–7.64 (m, 12H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 14.2 (×3), 22.8 (×3), 26.1, 26.4, 28.0 (×3), 28.7, 28.9 (×3), 29.5, 29.6 (×3), 29.7 (×5), 29.8, 30.2, 30.7, 32.0 (×3), 47.0, 54.0, 55.2, 55.3 (×2), 64.7, 67.3, 68.6, 71.8, 73.8, 74.6, 80.8, 81.8, 83.0, 97.9, 99.7, 113.9 (×2), 120.1 (×2), 125.2, 125.3, 127.2 (×2), 127.8 (×2), 129.9 (×2), 130.3, 141.4 (×2), 143.9 (×2), 155.9, 159.4, 168.4, 171.7, 172.0, 181.3 ppm; HR FAB MS [M+Na]+ calcd for C69H105NO14Na 1194.7433, found 1194.7433.Methyl 6-O-(4-((S)-2-(9-fluorenylmethoxycarbonyl)amino-2-carboxyethyl)oxy-4-oxobutanoyl)-2,3-di-O-tetradecanyl-α-D-glucopyranoside (24)
Conjugate 23 (0.2 g, 0.17 mmol) was dissolved in TFA/wet CH2Cl2 (1/5, v/v, 4 mL) and the resulting mixture was stirred for 2 h at rt. After that, the volatiles were evaporated under the reduced pressure, the residue was diluted with CH2Cl2 (5 mL) and neutralized with triethylamine (until pH ~ 7). The volatiles were removed under the reduced pressure and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane 1/1, v/v) to obtain the title compound 24 (0.13 g, 0.14 mmol) as a white amorphous solid in 81% yield. Analytical data for 24: Rf 0.58 (methanol/ethyl acetate, 1.5/8.5, v/v); [α]26D +34.9o (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3/(CD3)2SO, 2/1, v/v): δ, 0.78 (t, 6H, 2 × CH3), 1.45 (br. s, 40 H, 20 × CH2), 1.47 (m, 4H, 2 × CH2), 2.5 (m, 4H, 2 × CH2), 3.10 (dd, 1H, J2,3 = 9.4 Hz, H-2), 3.22–3.33 (m, 7H), 3.41–3.51 (m, 3H), 3.55 (m, 1H, H-5), 3.64 (t, 2H, J = 6.7 Hz), 4.14–4.32 (m, 7H), 4.42 (d, 1H, J = 9.5 Hz), 4.63 (d, 1H, J1,2 = 3.2 Hz,, H-1), 6.52 (br. s, 1H, Ser-NH), 7.22–7.82 (m, 8H, aromatic) ppm; 13C NMR (125 MHz, CDCl3/(CD3)2SO, 2/1, v/v): δ, 13.6 (×3), 21.9 (×3), 25.4 (×2), 25.5 (×2), 28.6 (×3), 28.8, 28.9 (×4), 29.0 (×5), 29.5, 31.2 (×3), 46.6, 54.3 (×2), 63.4, 64.9, 65.6, 69.2, 69.4, 70.6, 72.7, 79.6, 80.7, 97.3, 119.4 (×2), 124.6, 124.8, 126.6 (×2), 127.1 (×2), 140.1 (×2), 143.4 (×2), 155.4, 171.3, 171.6, 178.3, 178.7 ppm; HR FAB MS [M+Na]+ calcd for C57H89NO13Na 1018.6232, found 1018.6182.Preparation of compounds for cellular treatment
Synthetic compounds in solid form were dissolved in tetrahydrofuran (THF), aliquotted in small volumes, vacuum-centrifuged for 1 h and stored at −80 °C as dry compounds. For cellular treatment, an aliquot was dissolved in an appropriate volume of dimethylsulfoxide (DMSO) to give a concentrated working stock solution. This stock solution was further diluted in DMSO to give the desired concentration range. The final concentration of DMSO in the cell treatments was always maintained at 0.6%. Concentrations of Fmoc-containing compounds were verified by absorbance using an extinction coefficient of 7800 M−1cm−1 at 301 nm.Cell culture and LPS antagonism assays
THP-1 cells were obtained from ATCC (Manassas, VA) and maintained in a RPMI-1640 culture medium (HyClone, Logan, UT) containing 2 mM L-glutamine, 25 mM HEPES, 1.5 g L−1sodium bicarbonate, 10% fetal bovine serum (FBS) (HyClone), 50 units mL−1penicillin, 50 μg mL−1streptomycin (HyClone), and 50 μM β-mercaptoethanol at 37 °C in 5% CO2. For cellular assays, THP-1 monocytes were centrifuged and resuspended in a fresh growth medium to a cell density of 5 × 105cells mL−1. Cells were then seeded in a 48-well plate and differentiated into adherent macrophages by treatment with 10 ng mL−1phorbol 12-myristate 13-acetate (PMA) (Sigma) for 24 h at 37 °C in 5% CO2. The non-adherent cells were removed and the adherent cells were washed and replenished with reduced FBS growth medium. For the two wells set aside for calculating percent differentiation, the cells were washed with PBS (Hyclone) prior to removal by 0.25% trypsin-EDTA. Cells in a separate well were removed with 0.25% trypsin and counted under the microscope using a hemocytometer to determine the number of adherent macrophages. For the remaining wells, the cells were pre-incubated with LPS antagonist compounds at different concentrations for 30 min followed by the addition of 10 ng mL−1 ultrapure LPS from E. coliK12 (InvivoGen, San Diego, CA) for 6 h at 37 °C in 5% CO2. 0.6% DMSO was used as a control. The cell medium was collected and stored at −20 °C until analyzed by ELISA for secreted TNFα production.Cell viability measurements
Cell viability was monitored using an XTT [2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] assay. Macrophage metabolic activity was assessed by probing mitochondrial reduction of XTT (Sigma) which is a measure of viability (or toxicity) in response to the synthetic antagonists. Following 6 h treatment with antagonist compounds and LPS, the macrophages were washed with PBS (Hyclone) and incubated with XTT (0.33 mg mL−1) and phenazine methosulfate (PMS) (8.3 μM) (Acros, Morris Plains, NJ) to a final concentration of 0.33 mg mL−1 and 8.3 μM respectively for 2 h at 37 °C in 5% CO2. The cellular toxicity was then assessed in a platereader using absorbance of reduced XTT at 467 nm.Acknowledgements
This work was supported by awards from the AHA (0855743G to AVD) and NIH (1R15AG033913-01 to MRN). AFGB, MLDU, and SK are grateful to the UM—St. Louis Graduate School for awarding them with the Dissertation Fellowship in 2008, 2009, and 2010, respectively. Dr Winter and Mr. Kramer (UM—St. Louis) are thanked for HRMS determinations.References
- Gram-negative septicemia and septic shock, ed., L. Young and M. Glauser, WB Saunders, Philadelphia, 1991 Search PubMed.
- S. Tabbutt, Crit. Care Med., 2001, 29, S231–S236 CrossRef CAS.
- A. F. Suffredini, R. E. Fromm, M. M. Parker, M. Brenner, J. A. Kovacs, R. A. Wesley and J. E. Parrillo, N. Engl. J. Med., 1989, 321, 280–287 CrossRef CAS.
- U. Zahringer, B. Lindner and E. T. Rietschel, Adv. Carbohydr. Chem. Biochem., 1994, 50, 211–276 CrossRef CAS.
- C. R. H. Raetz, R. J. Ulevitch, S. D. Wright, C. H. Sibley, A. Ding and C. F. Nathan, FASEB J., 1991, 5, 2652–2660 CAS.
- T. A. Garrett and C. R. H. Raetz, Carbohydrates in Chemistry and Biology, ed. B. Ernst, G. W. Hart and P. Sinay, Wiley-VCH, Weinheim, New York, 2000, pp. pp. 435–447. Search PubMed.
- M. S. Jin and J. O. Lee, Immunity, 2008, 29, 182–191 CrossRef CAS.
- D. P. Rossignol, L. D. Hawkins, W. J. Christ, S. Kobayashi, T. Kawata, M. Lynn, I. Yamatsu and Y. Kishi, Endotoxin in Health and Disease, ed. H. Brade, S. M. Opal, S. N. Vogel and D. C. Morrison, Marcel Dekker, Inc., New York - Basel, 1999, pp. pp. 699–717. Search PubMed.
- W. J. Christ, P. D. McGuinness, O. Asano, Y. Wang, M. A. Mullarkey, M. Perez, L. D. Hawkins, T. A. Blythe, G. R. Dubuc and A. L. Robidoux, J. Am. Chem. Soc., 1994, 116, 3637–3638 CrossRef CAS.
- N. P. J. Price, B. Jeyaretnam, R. W. Carlson, J. L. Kadrmas, C. R. H. Raetz and K. A. Brozek, Proc. Natl. Acad. Sci., USA, 1995, 92, 7352–7356 CrossRef CAS.
- A. V. Demchenko, M. A. Wolfert, B. Santhanam, J. N. Moore and G. J. Boons, J. Am. Chem. Soc., 2003, 125, 6103–6112 CrossRef CAS.
- M. Shiozaki, H. Doi, D. Tanaka, T. Shimozato and S. Kurakata, Tetrahedron, 2006, 62, 205–225 CrossRef CAS.
- F. Peri, F. Granucci, B. Costa, I. Zanoni, C. Marinzi and F. Nicotra, Angew. Chem., Int. Ed., 2007, 46, 3308–3312 CrossRef CAS.
- H. G. Bazin, M. W. Wolff and R. J. Linhardt, J. Org. Chem., 1999, 64, 144–152 CrossRef CAS.
- M. Schultz and H. Kunz, Tetrahedron: Asymmetry, 1993, 4, 1205–1220 CrossRef CAS.
- L. Le Corre, C. Gravier-Pelletier and Y. Le Merrer, Eur. J. Org. Chem., 2007, 5386–5394 CrossRef CAS.
- W. J. Christ, O. Asano, A. L. C. Robidoux, M. Perez, Y. Wang, G. R. Dubuc, W. E. Gavin, L. D. Hawkins, P. D. McGuinness, M. A. Mullarkey, M. D. Lewis, Y. Kishi, T. Kawata, J. R. Bristol, J. R. Rose, D. P. Rossignol, S. Kobayashi, L. Hishinuma, A. Kimura, N. Asakawa, K. Katayama and I. Yamatsu, Science, 1995, 268, 80–83 CAS.
- M. Akamatsu, Y. Fujimoto, M. Kataoka, Y. Suda, S. Kusumoto and K. Fukase, Bioorg. Med. Chem., 2006, 14, 6759–6777 CrossRef CAS.
- K. Deng, M. M. Adams and D. Y. Gin, J. Am. Chem. Soc., 2008, 130, 5860–5861 CrossRef CAS.
- K. K. Maiti, M. DeCastro, A. B. M. A. A. El-Sayed, M. I. Foote, M. A. Wolfert and G. J. Boons, Eur. J. Org. Chem., 2010, 80–91 CAS.
- S. Tang, Q. Wang and Z. Guo, Chem.–Eur. J., 2010, 16, 1319–1325 CrossRef CAS.
- N. Moitessier, P. Englebienne and Y. Chapleur, Tetrahedron, 2005, 61, 6839–3853 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00145k |
| This journal is © The Royal Society of Chemistry 2011 |