Amine-linker length dependent electron transfer between porphyrins and covalent amino-modified single-walled carbon nanotubes
Konggang
Qu
ab,
Haixia
Xu
ab,
Chao
Zhao
ab,
Jinsong
Ren
a and
Xiaogang
Qu
*a
aDivision of Biological Inorganic Chemistry, State Key Laboratory of Rare Earth Resource Utilization, Laboratory of Chemical Biology, Changchun Institute of Applied Chemistry, Changchun, Jilin 130022, China. E-mail: xqu@ciac.jl.cn; Tel: +86 431 85262625
bGraduate School of the Chinese Academy of Sciences, Chinese Academy of Sciences, Changchun, Jilin 130022, China
First published on 23rd August 2011
Abstract
Single-walled carbon nanotube (SWNT)-porphyrin hybrid materials have received much attention due to their promising applications in photovoltaic devices, photodynamic therapy and energy conversion. Herein, three covalent amine-modified single-walled carbon nanotubes with different alkyl chain lengths have been synthesized and characterized by FT-IR, 1H NMR, XPS, and TGA-DTA methods. The electron transfer (ET) between SWNTs and porphyrins has been studied and the important influencing ET factors, such as linker length, solvent polarity, pH and ionic strength, and central metal of porphyrin, are discussed in detail in this report. Our results indicate that shorter alkyl chain length, higher polarity of the solvent, lower pH and ionic strength are all favorable for ET, the central metal coordinated to the porphyrin macrocycle is not essential for ET.
1. Introduction
As the leading nanodevice candidate, single-walled carbon nanotubes (SWNTs) are an attractive platform for the development of optoelectronic and photovoltaic devices.1–3 The presence of extended, delocalized π-electron systems makes them very useful for modulation of electron transfer when combined with photoexcited electron donors, such as porphyrins or metalloporphyrins.4–17 However, pristine SWNTs are difficult to disperse or dissolve in water and in organic media, and have the propensity to self-organize into densely packed hexagonal bundles or twisted ropes of nanotubes,18 which hamper their applications. Chemical modification and functionalization of SWNTs has made it possible to dissolve or disperse carbon nanotubes in water,19–22 thus opening the path for their facile manipulation and processing in physiological environments. Moreover, functionalized SWNTs (f-SWNTs) display lower toxicity than pristine SWNTs and can penetrate through the cell membrane and are not immunogenic.23–28 So, detailed studies of the electron transfer process between f-SWNTs and electron donors are required for their potential use in nanotechnology and bio-applications. And this might lead to novel, highly efficient photoelectrochemical cells.Porphyrins are stable natural functional dyes with a large extinction coefficient in the visible light region, predictable rigid structures, and prospective photochemical electron-transfer ability, and have been widely used for various photo-harvesting and photoelectronic devices.8,29–38 Since Nakashima and co-workers reported the first porphyrin–nanotube nanocomposite formed due to van der Waals forces,4 extensive SWNT–porphyrin hybrid materials have been investigated for various molecular photoelectronic applications.8–17 Guldi et al.26,33–38 have studied the interactions between SWNTs and porphyrin using diverse methods to fabricate SWNT–porphyrin hybrid systems, including covalent and noncovalent functionalization. However, detailed studies on different factors influencing the electron transfer between SWNTs and porphyrin have not been reported before.
In this report, SWNTs were first covalently functionalized with three diamines with different alkyl chain lengths,39,40 and the electron transfer between donor–acceptor self-assembled porphyrins or metalloporphyrins-SWNT nanohybrids has been studied (Scheme 1a) and the important influencing ET factors, such as linker length, solvent polarity, pH and ionic strength, central metal coordinated to the porphyrin macrocycle, are discussed.
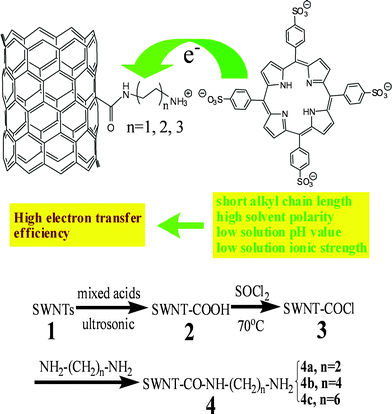 | ||
| Scheme 1 (a) Schematic illustration of the electron transfer between covalent amine-modified SWNTs and porphyrin. (b) Detailed procedures for the preparation of the amine functionalized SWNTs with different alkyl chain lengths. | ||
2 Experimental
2.1. Materials
SWNT (ϕ = 1.1 nm, purity > 90%) were purchased from Aldrich. Meso-tetra-(4-sulfonatophenyl) porphine dihydrochloride (H2TPPS), Cu(II) meso-tetra-(4-sulfonatophenyl) porphine dihydrochloride (CuTPPS) (acid form), meso-tetra-(4-carboxyphenyl) porphine dihydrochloride (H2TCPP) and Cu(II) meso-tetra-(4-carboxyphenyl) porphine dihydrochloride (CuTCPP) (acid form) were all obtained from Frontier Scientific Incorporation and used as received. The others are analytical or biochemical reagents.Functionalization: A similar procedure was adopted for the preparation of functionalized SWNTs 4 (Scheme 1b) as previously reported by Haddon,20 where the reactants were three terminal diamines instead of octadecylamine. Unlike the syntheses of 4a by treating 3 with ethylenediamine at room temperature, the synthesis of amide derivative 4b and 4c from 3 required heating at 80 °C and 100 °C with 1,4-butanediamine and 1,6-hexanediamine, respectively.
2.2. Apparatus and characterization
IR characterization was carried out on a BRUKE Vertex 70 FT-IR spectrometer; 1H NMR spectroscopy were recorded on a BRUKE AV400 (400 MHz) spectrometer using TMS (0 ppm) as the internal standard. Chemical shifts were reported in parts per million (ppm). X-ray photoelectron spectroscopy (XPS) spectra were obtained with an ESCALAB Thermal 250 instrument and monochromatic Mg-Kα (E = 1253.6 eV) was used for photoexcitation. TGA-DTA was performed on a SDT 2960 Thermal Gravity Analyzer, 5 mg of SWNTs derivatives 4 was placed in a TGA pan and heated up to 900 °C at a rate of 10 °C min−1 and flowed with dry nitrogen to detect the amount of terminal diamines attached to SWNTs.2.3. Sample preparation
The SWNTs derivatives 4 were dissolved in pure water at a concentration of 200 mg L−1. All solutions were sonicated for 30 min at room temperature in a water bath and briefly sonicated once again prior to use. The four porphyrin molecules used, the H2TPPS and CuTPPS, were dissolved in pure water to reach the concentration of 0.6 mM and the pH values of the porphyrin solutions were adjusted to ∼7.0 with freshly diluted ammonia; the H2TCPP and CuTCPP with poor water solubility were dissolved in DMSO and diluted in pure water to the concentration of 0.6 mM.2.4. Physical characterization of electron transfer between 4 and porphyrins
The electron transfer between 4 and porphyrins was characterized by UV-Vis spectra performed on a JASCO V-550 spectrophotometer in a 10 mm quartz cuvette and Fluorescence spectra on a JASCO FP-6500 fluorophotometer with slit widths of 10 nm for both excitation and emission monochromators in a 10 mm quartz cuvette. The UV-Vis spectra were measured by the addition of the same amount of suspensions of 4 into 1 μM (for sulfate porphyrins) or 2 μM (for carboxyl porphyrins, whose extinction coefficients are smaller) porphyrin solutions and the control solutions. The fluorescence titration experiments were carried out similar to the UV-Vis measurements without control solutions. In the fluorescence job plots, the total concentration of H2TPPS and 4 was 10 mg L−1.3. Results and discussion
3.1. Characterization of the functionalized SWNTs
Following Haddon's method, the as-prepared carboxyl modified SWNTs were used to attach the amine groups (Scheme 1b). First, the carboxyl modified SWNTs were treated with SOCl2 at 70 °C to convert the –COOH to –COCl groups. After separation, purification and dried under vacuum, the SWNT-COCl samples were reacted with excess ethylenediamine, 1,4-butanediamine, and 1,6-hexanediamine at room temperature, 80 °C and 100 °C, respectively. Followed by washing with ethanol and dichloromethane, the resulting black solid was dried at room temperature under vacuum. In contrast to the pristine SWNTs, which are insoluble in aqueous solutions, the as prepared products had a good solubility (> 200 mg L−1) in water. And the black-colored stock solution was very stable, almost no precipitation was observed upon prolonged standing under 4 °C.The FT-IR spectra of 2 and 4a indicate the formation of –COOH and the amide bond (Fig. 1a): (–COOH) νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1726 cm−1, νC–O = 1231 cm−1;19,39 (–CONH–) νCO = 1652 cm−1,3νC–N = 1212 cm−1, δN–H = 1550 cm−1.22Proton nuclear magnetic resonance (1H NMR, 400 MHz, D2O) spectrum of 4a shows the presence of the amide bond (Fig. 1b). The small sharp peak around 8 ppm corresponds to 1H of the amide bond, consistent with Hamers's report.40
O = 1726 cm−1, νC–O = 1231 cm−1;19,39 (–CONH–) νCO = 1652 cm−1,3νC–N = 1212 cm−1, δN–H = 1550 cm−1.22Proton nuclear magnetic resonance (1H NMR, 400 MHz, D2O) spectrum of 4a shows the presence of the amide bond (Fig. 1b). The small sharp peak around 8 ppm corresponds to 1H of the amide bond, consistent with Hamers's report.40
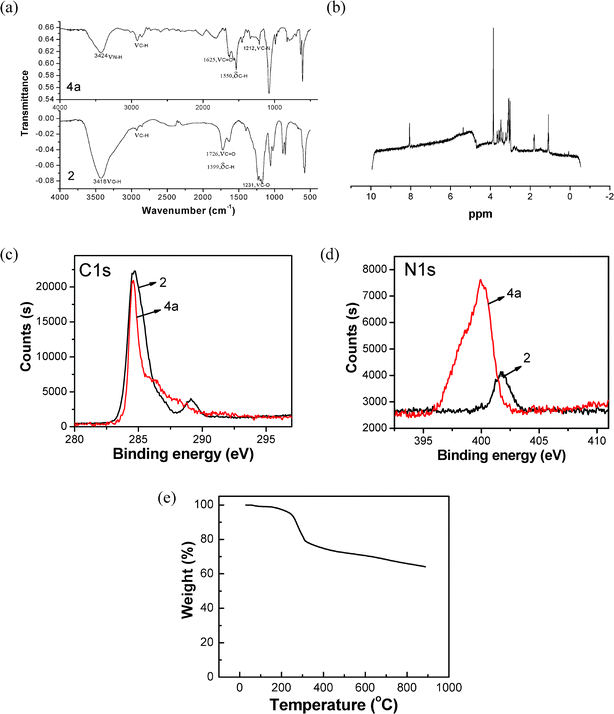 | ||
| Fig. 1 (a) FT-IR spectra of SWNT derivatives (A: 4a; B: 2). (b) 1H NMR spectrum of SWNT derivative 4a. (c–d) C 1s (c) and N 1s (d) XPS spectra of SWNT derivatives 2 (black line) and 4a (red line), respectively. (e) TGA spectrum of SWNT derivative 4a. | ||
In the C 1s (Fig. 1c) XPS spectrum of 2, there is a large peak at 284.7 eV from the nanotubes and a small peak at 289.1 eV assigned to carbonyl group of carboxylic acid group.40,41 However, in the spectrum of 4a, the peak around 285 eV became a little narrower, and the significant peak at 289 eV associated with carboxylic acid disappeared that were consistent with previous report.40 At the same time, the N 1s (Fig. 1d) spectra of 4a showed a broad peak with a binding energy of 399.9 eV. According to previous studies that amides and amines both have binding energies in the range of 399.5–400.5 eV,22,40 this indicates the formation of the amide bond, while the weak peak at 401.7 eV in the spectrum of SWNT-COOH may be attributed to the nitric acid brought by acids treatment.
In the TGA-DTA experiments, the weight loss in the temperature range of 150∼400 °C can be due to the degradation of the covalently tethered ethylenediamine (Fig. 1e) and the weight above 800 °C corresponds mainly to SWNTs,42 the small weight loss before 100 °C may be caused by physical adsorption of water. The amount of ethylenediamine, 1,4-butane diamine and 1,6-hexane diamine can be estimated 35.84, 36.22 and 42.69%, respectively.
3.2. Electron transfer (ET) between 4 and porphyrins
Since porphyrins have excellent photochemical properties, fluorescence and UV-vis spectral techniques were used to study the interactions between the amine-modified SWNTs and porphyrins. Fig. 2 shows porphyrin fluorescence spectral changes titrated by 4a. The intrinsic fluorescence intensity of H2TPPS (Fig. 2A) or H2TCPP (Fig. 2B) is quenched gradually as the 4a concentration increased. And the main difference between H2TPPS (Fig. 2A) and H2TCPP (Fig. 2B) is whether the fluorescence peak is shifted. Addition of 4a can decrease fluorescence intensity of H2TPPS (Fig. 2A) with no shift, while H2TCPP (Fig. 2B) has a slight red-shift from 642 nm to 647 nm. As reported previously, SWNTs can interact with porphyrin molecules through non-covalent π–π interactions, and they can form stable complexes.8–17,31–33 Therefore, fluorescence quenching of H2TPPS and H2TCPP indicates porphyrin binding to amine-modified SWNTs, and their interactions may even cause H2TCPP to be fluorescence shifted. | ||
| Fig. 2 Fluorescence titration of porphyrin molecules (A: H2TPPS; B: H2TCPP) with 4a (from 0 to 5 mg L−1). The concentrations of H2TPPS and H2TCPP are 1 μM and 2 μM, respectively. | ||
Generally, the interactions between SWNTs and porphyrins can be described: ground state charge transfer complex formation, after photo-excitation, followed by the electron transfer. Two factors may lead to the decrease in the quantum yield: intermolecular energy transfer or electron transfer. Previous studies have shown that the electron transfer between porphyrin and SWNTs is extremely sensitive to the polarity of the solvent.43–45 Thus the interactions of 4 and porphyrins in different solvents (water, methanol and DMF, whose dielectric constants are 80, 32.7 and 37.8, respectively) have been carried out, and the results fitted with Stern–Volmer equation
| F0/F = 1 + K × CSWNTs-NH2 | (1) |
 | ||
| Fig. 3 Relationship between fluorescence quenching constant changes of H2TCPP and the alkyl chain length of SWNTs derivatives in different polarity solvent (black: water; red: methanol; green: DMF). | ||
To corroborate that the occurrence of ET between 4 and porphyrins is attributed to the presence of SWNTs, we also studied the interaction between H2TPPS and ethylenediamine as a control. In fluorescence titration experiments, ethylenediamine causes little change of the fluorescence intensity of porphyrin (Fig. 4A) while 4a decreases the intensity significantly (Fig. 4B). In UV-vis titration experiments, ethylenediamine causes the Soret band of H2TPPS blue shifted without dramatically change of the absorbance (Fig. 4C), while 4a causes decrease of the absorbance and the Soret band blue-shifted (Fig. 4D). This further supports that H2TPPS can bind to the protonated amino group in either ethylenediamine or SWNT-NH2 by electrostatic interactions. However, SWNTs can decrease H2TPPS absorption indicating charge transfer complex formation between SWNTs and H2TPPS. After photo-excitation (Fig. 3), ET occurrs between H2TPPS and SWNTs.5
 | ||
| Fig. 4 Fluorescence (A and B) and UV-Vis (C and D) titration curves of H2TPPS against ethylenediamine (A and C) and SWNTs derivative of ethylenediamine 4a (B and D). | ||
Intriguingly, the alkyl linker length of the terminal diamine significantly influences ET between porphyrin and SWNTs (Fig. 3). Increasing alkyl chain length significantly decreased their ET. The plots of the quenching constants calculated by Stern–Volmer equation vs. the alkyl chain length of terminal diamines are shown in Fig. 5. Clearly, the ET efficiency decreased with the increase in alkyl chain length. This phenomenon can be explained well by the results obtained from absorbance measurements and the plots of the decrease of absorbance as a function of SWNT derivatives' concentration shown in Fig. 6. The SWNTs derivative having shorter alkyl chain length can cause porphyrin to take place more significant change in the absorbance, indicating stronger interactions and forming ground state charge transfer complex formation. This would promote following ET or stronger fluorescence quenching. Moreover, the effect of the alkyl chain length on the sulfate porphyrins is even stronger than that of carboxyl porphyrins.
 | ||
| Fig. 5 Relationship between fluorescence quenching constant and the alkyl chain length. | ||
 | ||
| Fig. 6 Absorbance changes of the four porphyrin molecules (A: H2TPPS; B: CuTPPS; C: H2TCPP; D: CuTCPP) with increasing the concentration of three SWNTs derivatives, respectively, (black: 4a; red: 4b; green: 4c); (E) relationship between the complex formation constants of the four porphyrin molecules and the alkyl chain length (black: H2TPPS; red: CuTPPS; green: H2TCPP; blue: CuTCPP). | ||
It is known that solution pH can influence the absorption of porphyrins. For H2TPPS (Fig. 7A), the Soret band is at 434 nm at pH 4, and shifts to 413 nm when pH > 5, meanwhile, the Soret band split into two peaks at pH 5. This is mainly caused by the existence of different forms of H2TPPS when the solution pH is changed, while solution pH has almost no effect on the absorbance of CuTPPS (Fig. 7B), which has no free H+. The effect of the pH on the ET efficiency between fluorescent porphyrins and SWNTs was investigated by fluorescence titration experiments (Fig. 8). For the two fluorescent porphyrins, it shows the highest ET efficiency at pH 5. Then the ET efficiency decreased gradually as the pH increased. This can be explained as follows: increasing solution pH would decrease protonation of SWNTs–NH2, and decrease the electrostatic interactions between 4 and porphyrin molecules.
 | ||
| Fig. 7 Effect pH on the absorption spectra of H2TPPS (A) and CuTPPS (B). | ||
 | ||
| Fig. 8 Effect of pH on the fluorescence quenching of the two porphyrins (A: H2TPPS; B: H2TCPP) with increase in the concentration of SWNT derivative 4a. The inset shows the relationship between quenching constants and solution pH. | ||
We also studied the effect of ionic concentration on the ET. Changing ionic concentration did not affect the Soret band of H2TPPS except small changes in the absorbance (Fig. 9A), however, for CuTPPS, changing ionic concentration can not only lead to the blue shift of the Soret band but also decreases the absorbance (Fig. 9B). For the two porphyrin molecules without central metal ions, fluorescence results show that increasing NaCl concentration also decreases their ET efficiency (Fig. 10). There are two main possible reasons for the ionic effect.48 One is cation ion, Na+, that can competitively bind to porphyrin molecules and would shield from the interactions with SWNTs-NH2; the other is the chloride ion, Cl−, that can exclude porphyrin from binding to positively charged SWNTs–NH2, these would slow the ET.48 Therefore, increasing ionic concentration decreases the electrostatic interactions between SWNTs-NH2 and porphyrin molecules.
 | ||
| Fig. 9 The absorption spectra of H2TPPS (A) and CuTPPS (B) in different NaCl concentration (from 0 to 1 M). | ||
 | ||
| Fig. 10 Ionic effect on the electron transfer between the two fluorescent porphyrins (A: H2TPPS; B: H2TCPP) and SWNT derivative 4a. The inset shows the relationship between quenching constants and NaCl concentration. | ||
The effect of the central metal coordinated to the porphyrin macrocycle was also studied. The results indicate that Cu as the central metal has a small effect on the ET efficiency between porphyrin and SWNTs derivatives in comparison with porphyrins without a coordinated central metal. This is inconsistent with previous reports that Zn as the central metal coordinated to porphyrin does not influence the dissolution of SWNTs.4
4. Conclusions
In summary, we have synthesized three diamine functionalized SWNTs with different alkyl chain lengths and the electron transfer between f-SWNTs and different porphyrin molecules was systematically studied. Our results indicate that shorter alkyl chain length of the terminal diamine, higher polarity of the solvent, lower pH value and ionic strength of the solutions are all favorable for electron transfer, the center metal coordinated to the porphyrin macrocycle is not essential for electron transfer. All these results may facilitate the design and synthesis of SWNT-based efficient electron donor–acceptor nanohybrid materials and their application in photovoltaic devices, photodynamic therapy and energy conversion.Acknowledgements
This project was supported by 973 Project (2011CB936004), NSFC (20831003, 90813001, 20833006, 90913007) and funds from the Chinese Academy of Sciences.References
- M. S. Dresselhaus, G. Dresselhaus and P. H. Avouris, Carbon Nanotubes: Synthesis, Structure, Properties, and Applications, New York, Springer Publishing, 2001 Search PubMed.
- S. Reich, C. Thomsen and J. Maultzsch, Carbon Nanotubes: Basic Concepts and Physical Properties, Germany, Weinheim, Wiley-VCH, 2004 Search PubMed.
- P. J. F. Harris, Carbon Nanotubes and Related Structures: New Materials for the Twenty-First Century. UK, Cambridge, Cambridge University Press, 2001 Search PubMed.
- H. Murakami, T. Nomura and N. Nakashima, Chem. Phys. Lett., 2003, 378, 481–485 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, J. Ramey, M. Marcaccio, D. Paolucci, F. Paolucci, S. Qin, W. T. Ford, D. Balbinot, N. Jux, N. Tagmatarchis and M. Prato, Chem. Commun., 2004, 2034–2035 RSC.
- D. M. Guldi, G. M. A. Rahman, M. Prato, N. Jux, S. Quin and W. Ford, Angew. Chem., Int. Ed., 2005, 44, 2015–2018 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, S. Quin, M. Tchoul, W. T. Ford, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli and M. Prato, Chem.–Eur. J., 2006, 12, 2152–2161 CrossRef CAS.
- D. Baskaran, J. W. Mays, X. P. Zhang and M. S. Bratcher, J. Am. Chem. Soc., 2005, 127, 6916–6917 CrossRef CAS.
- M. Alvaro, P. Atienzar, P. de la Cruz, J. L. Delgado, V. Troiani, H. García, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6626–6635 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, N. Jux, N. Tagmatarchis and M. Prato, Angew. Chem., Int. Ed., 2004, 43, 5526–5530 CrossRef CAS.
- H. Tanaka, T. Yajima, T. Matsumoto, Y. Otsuka and T. Ogawa, Adv. Mater., 2006, 18, 1411–1415 CrossRef CAS.
- D. S. Hecht, R. J. A. Ramirez, M. Briman, E. Artukovic, K. S. Chichak, J. F. Stoddart and G. Gruner, Nano Lett., 2006, 6, 2031–2036 CrossRef CAS.
- F. Cheng and A. Adronov, Chem.–Eur. J., 2006, 12, 5053–5059 CrossRef CAS.
- D. M. Guldi, H. Taieb, G. M. A. Rahman, N. Tagmatarchis and M. Prato, Adv. Mater., 2005, 17, 871–875 CrossRef CAS.
- C. Ehli, G. M. A. Rahman, N. Jux, B. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222–11231 CrossRef CAS.
- E. Maligaspe, A. S. D. Sandanayaka, T. Hasobe, O. Ito and F. D'Souza, J. Am. Chem. Soc., 2010, 132, 8158–8164 CrossRef CAS.
- M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, A. S. D. Sandanayaka, T. Hasobe and M. Yudasaka, Chem.–Eur. J., 2010, 16, 10752–10763 CrossRef CAS.
- K. S. Chichak, A. Star, M. V. R. Altoe and J. F. Stoddart, Small, 2005, 1, 452–461 CrossRef CAS.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y.-S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95–98 CrossRef CAS.
- H. Q. Peng, L. B. Alemany, J. L. Margrave and V. N. Khabashesku, J. Am. Chem. Soc., 2003, 125, 15174–15182 CrossRef CAS.
- T. Ramanathan, F. T. Fisher, R. S. Ruoff and L. C. Brinson, Chem. Mater., 2005, 17, 1290–1295 CrossRef CAS.
- D. Pantarotto, J. P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16–17 RSC.
- N. W. Shi Kam, T. C. Jessop, P. A. Wender and H. J. Dai, J. Am. Chem. Soc., 2004, 126, 6850–6851 CrossRef CAS.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS.
- S. Campidelli, C. Klumpp, A. Bianco, D. M. Guldi and M. Prato, J. Phys. Org. Chem., 2006, 19, 531–539 CrossRef CAS.
- L. Lacerda, A. Bianco, M. Prato and K. Kostarelos, Adv. Drug Delivery Rev., 2006, 58, 1460–1470 CrossRef CAS.
- H. Dumortier, S. Lacotte, G. Pastorin, R. Marega, W. Wu, D. Bonifazi, J. P. Briand, M. Prato, S. Muller and A. Bianco, Nano Lett., 2006, 6, 1522–1528 CrossRef CAS.
- T. S. Balaban, A. D. Bhise, M. Fischer, M. Linke-Schaetzel, C. Roussel and N. Vanthuyne, Angew. Chem., Int. Ed., 2003, 42, 2140–2144 CrossRef CAS.
- M. S. Choi, T. Yamazaki, I. Yamazaki and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 150–158 CrossRef CAS.
- Z. Guo, F. Du, D. Ren, Y. Chen, J. Zheng, Z. Liu and J. Tian, J. Mater. Chem., 2006, 16, 3021–3030 RSC.
- R. Chitta, A. S. D. Sandanayaka, A. L. Schumacher, L. D'Souza, Y. Araki, O. Ito and F. D'Souza, J. Phys. Chem. C, 2007, 111, 6947–6955 CrossRef CAS.
- B. Ballesteros, G. Torre, C. Ehli, G. M. A. Rahman, F. A. Rueda, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2007, 129, 5061–5068 CrossRef CAS.
- G. M. Aminur Rahman, D. M. Guldi, S. Campidellib and M. Prato, J. Mater. Chem., 2006, 16, 62–65 RSC.
- C. Ehli, G. M. Aminur Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222–11231 CrossRef CAS.
- S. Campidelli, C. Sooambar, E. L. Diz, C. Ehli, D. M. Guldi and M. Prato, J. Am. Chem. Soc., 2006, 128, 12544–12552 CrossRef CAS.
- S. Campidelli, B. Ballesteros, A. Filoramo, D. D. Díaz, G. de la Torre, T. Torres, G. M. Aminur Rahman, C. Ehli, D. Kiessling, F. Werner, V. Sgobba, D. M. Guldi, C. Cioffi, M. Prato and J.-P. Bourgoin, J. Am. Chem. Soc., 2008, 130, 11503–11509 CrossRef CAS.
- T. Palacin, H. L. Khanh, B. Jousselme, P. Jegou, A. Filoramo, C. Ehli, D. M. Guldi and S. Campidelli, J. Am. Chem. Soc., 2009, 131, 15394–15402 CrossRef CAS.
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180–192 CrossRef CAS.
- S. E. Baker, W. Cai, T. L. Lasseter, K. P. Weidkamp and R. J. Hamers, Nano Lett., 2002, 2, 1413–1417 CrossRef CAS.
- P. Y. Bruice, Organic Chemistry, Prentice Hall, 3rd edn, 2003, ch. 13, pp. 499 Search PubMed.
- G. X. Chen, H. S. Kim, B. H. Park and J. S. Yoon, J. Phys. Chem. B, 2005, 109, 22237–22243 CrossRef CAS.
- X. Qu, C. Wan, H-C. Becker, D. Zhong and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14212–14217 CrossRef CAS.
- H. P. Li, R. B. Martin, B. A. Harruff, R. A. Carino, L. F. Allard and Y. P. Sun, Adv. Mater., 2004, 16, 896–900 CrossRef CAS.
- M. Kozaki, K. Akita and K. Okada, Org. Lett., 2007, 9, 1509–1512 CrossRef CAS.
- X. Wang, C. Wang, K. Qu, Y. Song, J. Ren, D. Miyoshi, N. Sugimoto and X. Qu, Adv. Funct. Mater., 2010, 20, 3967–3971 CrossRef CAS.
- T. D. Gauthler, E. C. Shane, W. F. Guerln, W. R. Seltz and C. L. Grant, Environ. Sci. Technol., 1986, 20, 1162–1166 CAS.
- X. Li, Y. Peng, J. Ren and X. Qu, Biochemistry, 2006, 45, 13543–13550 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |