Novel optically active helical poly(N-propargylthiourea)s: synthesis, characterization and complexing ability toward Fe(III) ions†
Ci
Song
,
Lei
Li
,
Fangjie
Wang
,
Jianping
Deng
* and
Wantai
Yang
*
State Key Laboratory of Chemical Resource Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: dengjp@mail.buct.edu.cn; yangwt@mail.buct.edu.cn; Fax: +86-10-6443-5128; Tel: +86-10-6443-5128
First published on 21st October 2011
Abstract
An enantiomeric pair of novel chiral N-propargylthiourea monomers (monomers 1 and 2) and their racemate (monomer 3) were synthesized and polymerized in the presence of (nbd)Rh+B−(C6H5)4 as a catalyst. The resulting poly(1) and poly(2) could adopt stable helices and demonstrate optical activity in solvents of low polarity (e.g.CHCl3), according to circular dichroism and optical rotation measurements. But for poly(3), no considerable CD signal was observed at 320 nm and above. Poly(1) and poly(2) kept stable helical structures in a temperature range from 0–60 °C, while failed to maintain helix in polar solvents. Helical poly(1) and poly(2) exhibited strong complexing ability towards Fe(III) ions, based on which novel sensors may be developed next.
1. Introduction
Chiral macromolecules extensively occur in nature. More importantly, they adopt helical structures which play key roles for living organisms to maintain normal bioactivity. Inspired by naturally occurring biomacromolecules, various artificial polymers with chiral helical structures were successfully designed and prepared.1 Among these polymers, polyacetylenes are a typical class of synthetic helical polymers. Some groups2 have made remarkable contribution to acetylene-based chiral polymers, affording great progress in this significant research field. On the other hand, chiral thiourea and its derivatives have received a great deal of attention in particular because of the excellent asymmetric catalysis activity3 and intriguing metal-complexing ability.4 Jacobsen group5 prepared chiral thiourea-based catalysts to perform the asymmetric Strecker reactions of aldimines and ketoimines, which showed high yields and 99% enantiomeric excess. Takemoto group6 reported on chiral thiourea organocatalysts with functional amine groups that catalyzed enantioselective Michael reactions. Ganjali and coworkers7 developed a highly selective and sensitive Gd(III) membrane based on thiourea derivatives as an excellent neutral ion carrier. However, no reports can be found in literature focusing on introducing chiral thiourea moieties in polyacetylene backbones. A combination of functional thiourea groups and polyacetylene backbone will undoubtedly afford a variety of advanced functional materials.We have successfully prepared three categories of helical substituted polyacetylenes and their derivatives, i.e. poly(N-propargylamide)s,8 poly(N-propargylsulfamide)s,9 and poly(N-propargylurea)s.10 In our previous study, a series of achiral thiourea moieties were successfully introduced in polyacetylene backbones as side chains.11 Regretfully, the resulting polymers could not form helical conformation under the examined conditions. Based on the earlier poly(N-propargylthiourea)s without helices, two novel chiral poly(N-propargylthiourea)s (poly(1) and poly(2)) and an achiral poly(N-propargylthiourea) (poly(3)) were further designed and synthesized for the first time in this study. The obtained poly(1) and poly(2) could adopt helical structures, showing remarked positive or negative CD signals at around 320 nm in CD spectra. Furthermore, the two helical polymers showed strong complexing ability towards Fe(III) ions. The secondary structures of poly(1) and poly(2) dramatically changed after the adsorption of Fe(III) ions. The obtained results are thus of highly importance for further designing novel advanced functional materials such as chiral sensors, asymmetric catalysts, etc.
2. Experimental
Materials
All solvents were dried and distilled before use. Propargylamine (Aldrich), (R)-(+)-1-phenylethylamine, (S)-(−)-1-phenylethylamine, (±)-1-phenylethylamine, and p-toluenesulfonyl chloride (TsCl, Alfa Aesar) were used without further purification. Rhodium catalyst (nbd)Rh+B−(C6H5)4 (nbd = 2,5-norbornadiene) was synthesized according to the method reported in literature.12Measurements
FT-IR spectra were recorded with a Nicolet NEXUS 670 spectrophotometer (KBr tablet). The molecular weights (Mn) and molecular weight distributions (Mw/Mn) of polymers were determined by GPC (Waters 515–2410 system) calibrated by using polystyrene as standards and THF as eluent. 1H and 13C NMR spectra were recorded on a Bruker AV600 spectrometer. Elemental analysis was carried out with Thermo EA 1112 elemental analysis apparatus. Melting point (m.p.) was measured by an X-4 micro-melting point apparatus. Circular dichroism (CD) and UV-vis spectra were recorded on a JASCO J-810 spectropolarimeter at predetermined temperatures. Specific rotations were measured on a JASCO P-1020 digital polarimeter with a sodium lamp as the light source at room temperature.Synthesis of monomers
According to the earlier reference,13 a typical monomer synthesis procedure is as follows. Taking monomer 1 as an example, a 100 mL round bottomed flask was charged with (R)-(+)-1-phenylethylamine (1.5 mL, 11.7 mmol), triethylamine (Et3N, 5.0 mL, 36.0 mmol) and THF (15 mL), and then cooled with an ice bath under N2 atmosphere. Carbon disulfide (CS2, 0.70 mL, 11.7 mmol) was added to the reaction mixture by syringe over 0.5 h. After all the CS2 was added, the reaction mixture was stirred further at room temperature. 2 h later, the reaction mixture was cooled with an ice bath, and p-toluenesulfonyl chloride (TsCl, 2.3 g, 12.1 mmol) was added. The reaction system was allowed to spontaneously warm to room temperature. After 2 h, 1 N HCl (10 mL) and ethyl acetate (EtOAc, 10 mL) were added in the mixture. The aqueous layer was separated and back-extracted with EtOAc (10 mL). Then, the organic layers were collected and dried over anhydrous MgSO4. The product was filtered and concentrated to provide an oil, which was passed through a flash column chromatography on silica gel with 100% hexane as eluent. (R)-(+)-1-phenylethyl isothiocyanate (1.65 g) was obtained as a colorless oil in 86% yield. Next, (R)-(+)-1-phenylethyl isothiocyanate (0.53 g, 3.25 mmol) was dissolved in dried acetonitrile (20 mL). Then, 1.11 equiv. of propargylamine (0.25 mL, 3.63 mmol) was added dropwise into the isothiocyanate solution at 0 °C over 10 min. The solution was stirred for 24 h at room temperature. After that, the solution was concentrated to give the target monomer by evaporating the solvent. Finally, the crude product was further purified by flash column chromatography on silica gel (hexane/EtOAc = 1/1, v/v). Monomers 2 and 3 were synthesized in a similar way. The data for monomers 1–3 are as follows:Monomer 1: yield 62%, white solid, m.p. 83–84 °C. [a]D = 76° (c = 0.10 g dL−1, CHCl3). FT-IR (KBr): 3204, 3014 (NH), 2975 (–CH3), 1298, 1238 (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 693 (phenyl) cm−1. 1H NMR (CDCl3, 600 MHz, 20 °C): δ 1.52–1.53 (t, 3H, –CH3), 2.05 (s, 1H, H –C
S), 693 (phenyl) cm−1. 1H NMR (CDCl3, 600 MHz, 20 °C): δ 1.52–1.53 (t, 3H, –CH3), 2.05 (s, 1H, H –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ), 4.67 (d, 2H, –CH2 –C), 4.85 (d, 1H, –C(CH3)H–), 5.09–5.15 (s, 2H, –NH–CS–NH–), 7.26–7.35 (m, 5H, Ph–H). 13C NMR (CDCl3, 150 MHz, 20 °C): δ 23.17, 53.92, 66.79, 102.34, 125.97, 127.28, 128.63, 143.77, 148.69, 157.81. Calc. for C12H14N2S (218.09): C, 66.02; H, 6.46; N, 12.83; S, 14.69. Found: C, 66.17; H, 6.44; N, 12.77; S, 14.61.
), 4.67 (d, 2H, –CH2 –C), 4.85 (d, 1H, –C(CH3)H–), 5.09–5.15 (s, 2H, –NH–CS–NH–), 7.26–7.35 (m, 5H, Ph–H). 13C NMR (CDCl3, 150 MHz, 20 °C): δ 23.17, 53.92, 66.79, 102.34, 125.97, 127.28, 128.63, 143.77, 148.69, 157.81. Calc. for C12H14N2S (218.09): C, 66.02; H, 6.46; N, 12.83; S, 14.69. Found: C, 66.17; H, 6.44; N, 12.77; S, 14.61.
Monomer 2: yield 65%, white solid, m.p. 83–84 °C. [a]D = −78° (c = 0.10 g dL−1, CHCl3). FT-IR (KBr): 3201, 3010 (NH), 2980 (–CH3), 1301, 1236 (N –CS), 690 (phenyl) cm−1. 1H NMR (CDCl3, 600 MHz, 20 °C): δ 1.52–1.53 (t, 3H, –CH3), 2.05 (s, 1H, H –C), 4.67 (d, 2H, –CH2 –C), 4.85 (d, 1H, –C(CH3)H–), 5.09–5.14 (s, 2H, –NH –CS–NH–), 7.26–7.35 (m, 5H, Ph–H). 13C NMR (CDCl3, 150 MHz, 20 °C): δ 23.07, 53.84, 66.84, 102.44, 125.96, 127.33, 128.65, 143.62, 148.57, 157.52. Calc. for C12H14N2S (218.09): C, 66.02; H, 6.46; N, 12.83; S, 14.69. Found: C, 66.15; H, 6.43; N, 12.74; S, 14.66.
Monomer 3: yield 60%, white solid, m.p. 82–83 °C. [a]D = 0° (c = 0.10 g dL−1, CHCl3). FT-IR (KBr): 3210, 3010 (NH), 2982 (–CH3), 1299, 1234 (N–CS), 688 (phenyl) cm−1. 1H NMR (CDCl3, 600 MHz, 20 °C): δ 1.52–1.53 (t, 3H, –CH3), 2.05 (s, 1H, H –C), 4.67 (d, 2H, –CH2 –C), 4.85 (d, 1H, –C(CH3)H–), 5.09–5.15 (s, 2H, –NH –CS–NH–), 7.26–7.35 (m, 5H, Ph–H). 13C NMR (CDCl3, 150 MHz, 20 °C): δ 23.12, 53.86, 66.86, 102.38, 125.97, 127.31, 128.64, 143.70, 148.66, 157.61. Calc. for C12H14N2S (218.09): C, 66.02; H, 6.46; N, 12.83; S, 14.69. Found: C, 66.07; H, 6.42; N, 12.85; S, 14.66.
Synthesis of poly(N-propargylthiourea)s
Referring to the earlier studies,11,14polymerizations were carried out with (nbd)Rh+B−(C6H5)4 as a catalyst in a dry dimethyl formamide (DMF) under N2 atmosphere, [monomer]0 = 0.5 M, [catalyst] = 5 mM. After polymerization, the resulting solution was put into vacuum oven for 24 h at 30 °C to evaporate the solvent. Then, 1 mL of chloroform was added into the residual and the mixture was poured into a large amount of hexane to precipitate the formed polymer. Finally, the polymer was filtered off, washed with hexane, and dried under reduced pressure. 1H and 13C NMR spectra of poly(1), which is taken as a representative, are shown in Fig. S1 (ESI).†3. Results and discussion
Synthesis and polymerization of N-propargylthiourea monomers
Monomers 1, 2, and 3 were synthesized for the first time, and then polymerized in the presence of (nbd)Rh+B−(C6H5)4 as a catalyst (Scheme 1). The details for monomer synthesis and polymerization procedures are described in detail in the experimental section. Polymerization results are summarized in Table 1.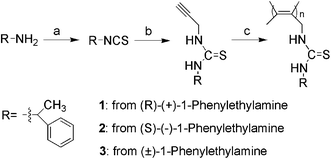 | ||
| Scheme 1 Monomer synthesis and polymerization. Reagents and conditions: (a) Et3N, CS2, TsCl, ice bath, N2 atmosphere, THF; (b) propargylamine, room temperature, dried acetonitrile; (c) (nbd)Rh+B−(C6H5)4, 30 °C, 6 h, DMF, N2 atmosphere. | ||
| Monomer | Polymer | |||||
|---|---|---|---|---|---|---|
| Entry | [α]D (deg)b | / | Yield (%)c | M n d | M w/Mnd | [α]D (deg)b |
| a With (nbd)Rh+B−(C6H5)4 as catalyst; [M]0 = 0.5 M; [M]0/[Rh] = 100; polymerizationT = 30 °C; in DMF for 6 h. b Measured by polarimeter at room temperature, c = 0.1 g dL−1 in CHCl3. c Hexane-insoluble part. d Measured by GPC (polystyrene as the standards; THF as eluent). | ||||||
| Monomer 1 | 76 | poly(1) | 73 | 3000 | 1.31 | 574 |
| Monomer 2 | −78 | poly(2) | 72 | 3100 | 1.34 | −586 |
| Monomer 3 | 0 | poly(3) | 69 | 2900 | 1.28 | 0 |
Table 1 demonstrates that all the monomers 1–3 underwent polymerizations smoothly and provided the corresponding polymers, i.e. poly(1)–poly(3) with a relatively high yield of ca. 70%. The polymers had similar molecular weights (Mn) about 3,000 (polymerization degree: approx. 14) and molecular weight distribution (Mw/Mn) about 1.30. It seems that the thiourea groups are not favorable for the monomers to form polymers with high molecular weight.11 As we expected, poly(1) and poly(2) showed high specific rotations compared with those of the corresponding monomers (Table 1), reflecting that both of them were optically active. However, poly(3) and monomer 3 did not show optical activity because they are short of chiral carbon atom in their chemical structure. More importantly, the relatively large specific optical rotations of poly(1) and poly(2) followed the well-studied “chiral amplification”1h,15 effect. This observation together with the CD spectra (presented below) indicate that poly(1) and poly(2) formed helical structures of predominant one-handedness, as to be discussed in more detail later on.
The solubilities of the three polymers were explored in several solvents. We found all the polymers were totally soluble in DMF, DMSO, THF, CHCl3, and CH2Cl2, but hardly soluble in toluene and ether.
Secondary structure of poly(N-propargylthiourea)s
Secondary structure of poly(1) was studied by measuring UV-vis and CD spectra, as shown in Fig. 1, since these techniques have been proved highly efficient for analyzing the secondary structures of substituted polyacetylenes.16 For a clear comparison, monomer 1 was also examined. For monomer 1, there is no considerable adsorption peak at a wavelength over 300 nm, but a strong absorption could be observed around 240 nm (in Fig. 1a). This peak is attributed to the thiourea and phenethyl groups in the monomer. In Fig. 1b, it can be seen that a slight positive CD signal appeared around 275 nm which is caused by the chiral carbon atom in monomer 1. For poly(1), there is an obvious adsorption peak at around 320 nm in UV-vis spectrum (Fig. 1a). Correspondingly, a strong positive CD signal appeared at 320 nm in CD spectra (Fig. 1b). Similar situations occurred in poly(2) and monomer 2. Nevertheless, they demonstrated negative CD signals in Fig. 2.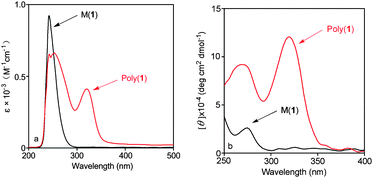 | ||
| Fig. 1 (a) UV-vis absorption and (b) CD spectra of monomer 1 and poly(1) (c = 0.1 mM, CHCl3, room temperature). | ||
 | ||
| Fig. 2 (a) UV-vis absorption and (b) CD spectra of monomer 2 and poly(2) (c = 0.1 mM, CHCl3, room temperature). | ||
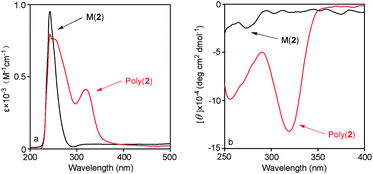
Achiral monomer 3 was investigated as a reference for revealing the secondary structures of the two chiral poly(N-propargylthiourea)s. The UV-vis and CD spectra of poly(1)–poly(3) are all depicted in Fig. 3 for a more clear comparison. All three polymers showed obvious UV-vis adsorption around 320 nm in Fig. 3a, however, the CD spectra in Fig. 3b are totally different. Poly(1) showed an obvious positive CD signal at 320 nm, and a corresponding negative CD signal was observed at the nearly identical wavelength for poly(2). Poly(3) did not contain any chiral centers, and just as expected, no CD signal was observed in its CD spectrum in 300–400 nm. Monomers 1 and 2 are a pair of enantiomers, i.e. R-(+) and S-(−) isomers, respectively, while monomer 3 is racemic. Therefore poly(1) and poly(2) exhibited stronger CD signals with positive and negative Cotton effect in their CD spectra, and poly(3) showed no CD signal. Referring to our earlier reports17 and based on the CD spectra and specific rotations in Table 1, it can be inferred that both poly(1) and poly(2) formed helical structures of predominant one-handed screw type and thus exhibited optical activities, while poly(3) formed helical conformations of both right- and left-handedness in equal amount.
 | ||
| Fig. 3 (a) UV-vis absorption and (b) CD spectra of poly1–poly3 (c = 0.1 mM, CHCl3, room temperature). | ||
For obtaining deep insights into the novel chiral poly(N-propargylthiourea)s, FT-IR spectroscopic measurements were carried out in solution. Herein, we took poly(1) and monomer 1 as typical examples. As shown in Fig. 4, 2122 cm−1 indicates the CC group. The absorption peaks at 3345, 1623 and 1056 cm−1 in the FT-IR spectrum of monomer 1 belonged to the stretching and bending vibration of N–H and the stretching vibration of CS, respectively. Compared with monomer 1, the FT-IR peaks of poly(1) shifted toward lower wavenumbers which appeared at 3273, 1608 and 1052 cm−1, respectively. These results demonstrated that thiourea moieties formed hydrogen bonding between CS and N–H groups. Moreover, the hydrogen bonding occurred intramolecularly because increasing or decreasing the concentration of poly(1) (15 and 60 mM) caused little change in the wavenumbers of the FT-IR peaks. Referring to our earlier studies,17,18 the hydrogen bonding is vital for the polymer main chains to adopt stable helical structures.
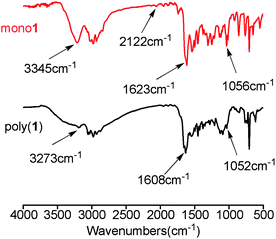 | ||
| Fig. 4 FT-IR spectra of monomer 1 and poly(1) measured in CHCl3 (c ≈ 30 mM, room temperature). | ||
Solvent effects
According to earlier literature,19solvent is a key factor for the secondary structure of helical polymers. DMF may destroy the hydrogen bonding in the neighboring side chains. To obtain more information on the helical conformation formed in poly(1) and poly(2), UV-vis and CD spectra of them were further measured in the mixture solvent consisting of CHCl3 and DMF. The results are illustrated in Fig. 5 and 6.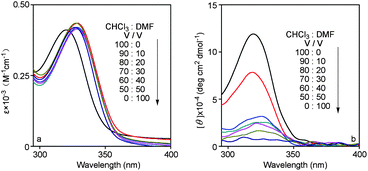 | ||
| Fig. 5 Solvent dependence of (a) UV-vis absorption and (b) CD spectra of poly(1) (c = 0.1 mM, room temperature). | ||
 | ||
| Fig. 6 Solvent dependence of (a) UV-vis absorption and (b) CD spectra of poly(2) (c = 0.1 mM, room temperature). | ||
In Fig. 5, the CHCl3/DMF composition varied from 100/0 to 0/100 (v/v). At room temperature in CHCl3, poly(1) exhibited a UV-vis absorption peak and a positive CD signal at around 320 nm. When a small amount of DMF (10%) was added into the solution of poly(1) in CHCl3, a red shift was observed in the UV-vis absorption spectra at about 328 nm, and the intensity of CD signal decreased. Then, the UV-vis absorption peaks of poly(1) little changed with increasing of amount of DMF. However, the CD signals almost disappeared when the content of DMF was increased up to 50%. Poly(1) in this case of 50% DMF took random coil structure without optically active because DMF destroyed the intramolecular hydrogen bonding in the side chains between N–H and CS groups. The same situation could be seen for poly(2) in Fig. 6. Thus, it can be concluded that CHCl3 was favorable for the formation of helical structure of poly(N-propargylthiourea)s, and the addition of DMF transformed the conformations of polymers from ordered helix to random coil. In addition, the dependence of the UV-vis absorption and CD signal on temperature of poly(1) and poly(2) were explored also, as shown in Fig. S2 and S3.† When the temperature was elevated from 0 to 60 °C, the UV-vis absorption changed little, while the CD signals weakened noticeably, indicating that racemization took place in the polymers to some degree upon increasing temperature.
Metal ion effects
Based on our previous study,11 poly(N-propargylthiourea)s without helical conformation exhibited considerable adsorption ability toward Fe(III) ions. In this work, helical polymers, i.e. poly(1) and poly(2) were used for forming metal complex with Fe(III) ions. The UV-vis absorption and CD spectra of poly(1) before and after adsorption of Fe(III) are shown in Fig. 7. For poly(1), there was a UV-vis absorption peak and a positive CD signal at around 320 nm. For the metal complex, with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio of poly(1) toward Fe(III) ions, the increasing of absorption peak at 320 nm can be ascribed to the coordination between thiourea groups and Fe(III) ions. The emerging absorption band at about 360 nm in UV-vis spectrum is caused by the introduction of Fe(III) ions. Moreover, the positive CD signal of poly(1) at around 320 nm totally disappeared after adsorption Fe(III). It indicated that the Fe(III) complex which formed between chiral polymers and Fe(III) could not take helical structure since the intramolecular hydrogen bonds were destroyed by the coordination between Fe(III) and thiourea groups in polymer pendent chains. A similar situation occurred in poly(2) and its Fe(III) complex, as shown in Fig. 8. According to the above results, this kind of chiral poly(N-propargylthiourea)s are expected to find remarkable applications in the area of metal ions detectors, chemical sensors, chiral catalysts, etc. The relevant work is in progress in our group.
:1 mol ratio of poly(1) toward Fe(III) ions, the increasing of absorption peak at 320 nm can be ascribed to the coordination between thiourea groups and Fe(III) ions. The emerging absorption band at about 360 nm in UV-vis spectrum is caused by the introduction of Fe(III) ions. Moreover, the positive CD signal of poly(1) at around 320 nm totally disappeared after adsorption Fe(III). It indicated that the Fe(III) complex which formed between chiral polymers and Fe(III) could not take helical structure since the intramolecular hydrogen bonds were destroyed by the coordination between Fe(III) and thiourea groups in polymer pendent chains. A similar situation occurred in poly(2) and its Fe(III) complex, as shown in Fig. 8. According to the above results, this kind of chiral poly(N-propargylthiourea)s are expected to find remarkable applications in the area of metal ions detectors, chemical sensors, chiral catalysts, etc. The relevant work is in progress in our group.
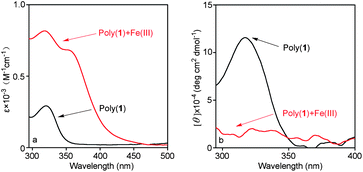 | ||
| Fig. 7 (a) UV-vis absorption and (b) CD spectra of poly(1) and Fe(III) complexes (1:1 mole ratio of poly(1) to Fe(III) ions). (c = 0.10 mM, CHCl3/DMF = 9:1(v/v), room temperature). | ||
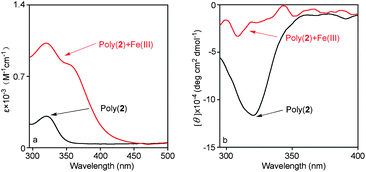 | ||
| Fig. 8 (a) UV-vis absorption and (b) CD spectra of poly(2) and Fe(III) complexes (1:1 mole ratio of poly(2) to Fe(III) ions). (c = 0.10 mM, CHCl3/DMF = 9:1(v/v), room temperature). | ||
4. Conclusions
In this report, an enantiomeric pair of novel chiral N-propargylthiourea monomers (monomers 1 and 2) and racemic monomer 3 were successfully synthesized and polymerized. The resulting poly(1) and poly(2) adopted helical structure of predominantly one-handedness and thus showed optical activity, while poly(3) formed helical conformations of both right- and left-handedness in equal amount under the examined conditions. The formation of helices exibited high dependence on the polarity of solvent. The addition of DMF destroyed the hydrogen bonds which formed between the neighboring thiourea groups. In addition, both two chiral polymers could not take helical structure after adsorption Fe(III) because the intramolecular hydrogen bonds were destroyed by the coordination between Fe(III) and thiourea groups in polymer pendent chains. Since this novel type of optically active helical polymers combines the conjugated polymeric main chains and the highly functional thiourea groups, we can expect interesting properties from them. The related investigations are in progress in our lab at present.Acknowledgements
This work is supported by the “Program for New Century Excellent Talents in University” (NCET-06-0096), the “National Natural Science Foundation of China” (21174010, 20974007), the “Fundamental Research Funds for the Central Universities” (ZZ1117), and the “Major Project for Polymer Chemistry and Physics Subject”.Notes and references
- Some representative articles: (a) J. G. Kennemur, J. B. Clark, G. Tian and B. M. Novak, Macromolecules, 2010, 43, 1867 CrossRef CAS; (b) T. Sakamoto, Y. Fukuda, S. Sato and T. Nakano, Angew. Chem., Int. Ed., 2009, 48, 9308 CrossRef CAS; (c) M. Fujiki, Top. Curr. Chem., 2008, 284, 119 CrossRef CAS; (d) J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641 CrossRef CAS; (e) M. Shiotsuki, F. Sanda and T. Masuda, Polym. Chem., 2011, 2, 1044 RSC; (f) T. Aoki, T. Kaneko, N. Maruyama, A. Sumi, M. Takahashi, T. Sato and M. Teraguchi, J. Am. Chem. Soc., 2003, 125, 6346 CrossRef CAS; (g) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS; (h) M. M. Green, J. W. Park, T. Sato, K. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138 CrossRef.
- Some representative articles: (a) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136 CrossRef CAS; (b) E. Yashima, K. Maeda, H. Iika, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS; (c) J. Liu, J. E. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS; (d) K. Akagi and T. Mori, Chem. Rec., 2008, 8, 395 CrossRef CAS; (e) V. Percec, B. C. Won, M. Peterca and P. A. Heiney, J. Am. Chem. Soc., 2007, 129, 11265 CrossRef CAS; (f) T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165 CrossRef CAS.
- (a) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030 CrossRef CAS; (b) Y. Zhang, Y. L. Shao, H. S. Xu and W. Wang, J. Org. Chem., 2011, 76, 1472 CrossRef CAS.
- T. Mandal and C. G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714 CrossRef CAS.
- P. Vachal and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 10012 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS.
- M. R. Ganjali, P. Norouzi, T. Alizadeh, A. Tajarodi and Y. Hanifehpour, Sens. Actuators, B, 2007, 120, 487 CrossRef.
- (a) L. Ding, Y. Y. Huang, Y. Y. Zhang, J. P. Deng and W. T. Yang, Macromolecules, 2011, 44, 736 CrossRef CAS; (b) Y. Y. Zhang, X. F. Luo, J. P. Deng and W. T. Yang, Macromol. Chem. Phys., 2011, 212, 353 CAS; (c) K. Zhou, L. Y. Tong, J. P. Deng and W. T. Yang, J. Mater. Chem., 2010, 20, 781 RSC; (d) L. Ding, X. F. Jiao, J. P. Deng, W. G. Zhao and W. T. Yang, Macromol. Rapid Commun., 2009, 30, 120 CrossRef CAS.
- (a) B. Chen, J. P. Deng and W. T. Yang, Colloid Polym. Sci., 2011, 289, 133 CrossRef CAS; (b) B. Chen, J. P. Deng and W. T. Yang, Macromolecules, 2010, 43, 9613 CrossRef CAS; (c) B. Chen, J. P. Deng, X. Q. Liu and W. T. Yang, Macromolecules, 2010, 43, 3177 CrossRef CAS; (d) J. P. Deng, B. Chen, X. F. Luo and W. T. Yang, Macromolecules, 2009, 42, 933 CrossRef CAS.
- (a) X. F. Luo, J. P. Deng and W. T. Yang, Angew. Chem., Int. Ed., 2011, 50, 4909 CrossRef CAS; (b) X. F. Luo, L. Li, J. P. Deng, T. T. Guo and W. T. Yang, Chem. Commun., 2010, 46, 2745 RSC.
- L. Li, X. F. Luo, X. Chang, J. P. Deng and W. T. Yang, Des. Monomers Polym., 2011, 14, 143 CrossRef CAS.
- R. R. Schrock and J. A. Osborn, Inorg. Chem., 1970, 9, 2339 CrossRef CAS.
- R. Wong and S. J. Dolman, J. Org. Chem., 2007, 72, 3969 CrossRef CAS.
- M. Tabata, T. Sone and Y. Sadahiro, Macromol. Chem. Phys., 1999, 200, 265 CrossRef CAS.
- (a) A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS; (b) K. Morino, N. Watase, K. Maeda and E. Yashima, Chem.–Eur. J., 2004, 10, 4703 CrossRef CAS , and references therein.
- A typical article: R. Nomura, Y. Fukushima, H. Nakako and T. Masuda, J. Am. Chem. Soc., 2000, 122, 8830 CrossRef CAS.
- (a) J. P. Deng, X. F. Luo, W. G. Zhao and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4112 CrossRef CAS; (b) Z. G. Zhang, J. P. Deng, W. G. Zhao, J. M. Wang and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 500 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 1891 CrossRef CAS.
- R. Nomura, J. Tabei and T. Masuda, Macromolecules, 2002, 35, 2955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00457c |
| This journal is © The Royal Society of Chemistry 2011 |