Functionalized polycarbonates from dihydroxyacetone: insights into the immortal ring-opening polymerization of 2,2-dimethoxytrimethylene carbonate†
Marion
Helou
,
Jean-Michel
Brusson
,
Jean-François
Carpentier
and
Sophie M.
Guillaume
*
Laboratoire Catalyse et Organométalliques, CNRS - Université de Rennes 1 - Sciences Chimiques de Rennes (UMR 6226), Campus de Beaulieu, 35042, Rennes Cedex, France. E-mail: sophie.guillaume@univ-rennes1.fr; Fax: +33 2 2323 6939; Tel: +33 2 2323 5880
First published on 14th October 2011
Abstract
Functionalized polycarbonates derived from 2,2-dimethoxytrimethylene carbonate (TMC(OMe)2) have been prepared with controlled molecular features by immortal ring-opening polymerization, under mild conditions (bulk, 60–90 °C), using various (metallo)organic/alcohol (diol) binary catalyst systems: the β-diiminate zinc complex [(BDIiPr)Zn(N(SiMe3)2)] (BDI = CH(CMeNC6H3-2,6-iPr2)2), the aluminium triflate, or the organic bases 4-N,N-dimethylaminopyridine (DMAP), 1.5.7-triazabicyclo-[4.4.0]dec-5-ene (TBD) and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP), as catalyst precursors, combined with benzyl alcohol or 1,3-propanediol acting both as a co-initiator and a chain transfer agent. For the first time, well-defined α-hydroxy-ω-alkoxyester and α,ω-dihydroxy telechelic acetal-functionalized homopolycarbonates were thus prepared with molar mass up to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1. These polymers were characterized at the molecular (NMR, SEC), thermal (DSC, TGA) and mechanical levels, and compared to conventional PTMC. P(TMC(OMe)2) is a rigid and brittle polymer material (E = 3190 ± 70 MPa, εr = 9 ± 1%).
200 g mol−1. These polymers were characterized at the molecular (NMR, SEC), thermal (DSC, TGA) and mechanical levels, and compared to conventional PTMC. P(TMC(OMe)2) is a rigid and brittle polymer material (E = 3190 ± 70 MPa, εr = 9 ± 1%).
Introduction
Synthetic biodegradable polycarbonates are receiving a topical burst of interest, especially in regard to their great versatility imparted by their tunable physico-chemical and notably thermo-mechanical properties, which make them highly valuable, non-toxic polymer materials. Polycarbonates are targeted in medical and pharmaceutical industries as biocompatible drug delivery systems, tissue repair or tissue regeneration biomaterials. Agricultural, microelectronics and packaging applications are also exploiting and benefiting from their biodegradability, aiming at diversified purposes ranging from specialties to commodities.1Polycarbonates bearing pendant functionalities along the backbone chain afford the advantage of tailoring the polymer physico-chemical properties to specific needs, thereby broadening and improving their performance characteristics. These can be synthesized either upon direct polymerization of the carefully designed functionalized monomer or upon post-polymerization chemical modification, the former direct polymerization approach being more attractive. Parameters such as solubility, hydrophilicity/hydrophobicity balance, crystallinity, degradation/resorption, permeability, and thermal and mechanical properties can thus be finely modulated. In addition, the side-chain functionalities may serve as further chemical modification sites or as anchoring spots for covalent attachment of e.g. pro-drugs.1,2
In order to address environmental issues, polymers nowadays have to be developed within “green” and sustainable considerations through bio-friendly synthetic approaches. In this regard, we have been (i) valorizing the biomass and (ii) extending ring-opening polymerization (ROP) processes of cyclic esters and carbonates to efficient atom-economic “immortal” procedures, for the controlled elaboration of polyesters/polycarbonates.3 Bio-resourced itaconic or levulinic acids4 as well as glycerol5 allowed the preparation of seven- and six-membered cyclic carbonates, respectively. Polycarbonates, especially poly(trimethylene carbonate)s (PTMCs), along with other polyesters such as poly(3-hydroxybutyrate)s, have been prepared upon “immortal” ROP (iROP) using several binary catalyst systems based on an alcohol and a metal- or organo-catalyst.3–6 We have now extended our investigations to 2,2-dimethoxytrimethylene carbonate (2,2-dimethoxypropane-1,3-diol carbonate, hereafter referred to as TMC(OMe)2), which is derived from naturally occurring 1,3-dihydroxyacetone (DHA) (Scheme 1).5e
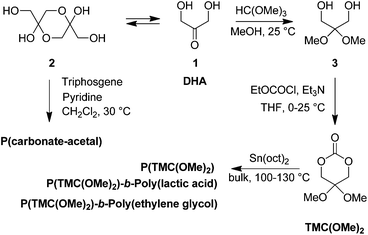 | ||
| Scheme 1 Reported syntheses of (co)polymers derived from TMC(OMe)2.7,8 | ||
DHA, an intermediate in the glucose metabolism derived from plant sources (sugar beets and sugar cane) and obtained directly by oxidation of glycerol, is biologically accepted by the human body and then most widely used as an active ingredient in sunless tanning cosmetics. The solution equilibrium of monomeric DHA (1) with its hemiacetal dimer (2) has been locked in the dimer form, allowing the preparation of poly(carbonate acetal)s (Scheme 1).7 Alternatively, DHA locked into its monomeric form through conversion of its C2 carbonyl into a dimethoxy acetal 3, led to the formation of TMC(OMe)2.8Polycarbonates obtained from the bulk ROP of TMC(OMe)2 at 100 °C using stannous octanoate (Sn(oct)2; no initiator specifically mentioned) have thus been scarcely described, yet quite briefly and with only three homopolymers of molar mass up to 37500 g mol−1 (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w/n ≤ 1.5).8 Indeed, the aim of these studies was rather the deprotection of the dimethyl acetal group toward the formation of poly(2-oxypropylene carbonate) to ultimately foster the polymer's surface functionalization through reductive amination of the C2 carbonyl, a nucleophilic addition at the carbonyl without the use of condensation reagents (Scheme 1).8aDiblock copolymers from DHA and ethylene glycol or lactide have also been reported, yet rarely, for amphiphilic nanoparticulate drug delivery vehicles' outcomes (Scheme 1).8b,c
w/n ≤ 1.5).8 Indeed, the aim of these studies was rather the deprotection of the dimethyl acetal group toward the formation of poly(2-oxypropylene carbonate) to ultimately foster the polymer's surface functionalization through reductive amination of the C2 carbonyl, a nucleophilic addition at the carbonyl without the use of condensation reagents (Scheme 1).8aDiblock copolymers from DHA and ethylene glycol or lactide have also been reported, yet rarely, for amphiphilic nanoparticulate drug delivery vehicles' outcomes (Scheme 1).8b,c
In this work, we report the ROP of TMC(OMe)2 under “immortal” conditions using various binary catalyst systems based on a metal- or organo-catalyst and an alcohol/diol acting as a co-initiator and a chain transfer agent (Scheme 2).9 The catalytic systems were selected among the most active ones in the ROP of the parent trimethylene carbonate (TMC) monomer.3–5 The polycarbonates were then molecularly, thermally and mechanically characterized.
![Schematic representation of the [catalyst/alcohol]-mediated iROP of TMC(OMe)2: synthesis of P(TMC(OMe)2).](/image/article/2011/PY/c1py00405k/c1py00405k-s2.gif) | ||
| Scheme 2 Schematic representation of the [catalyst/alcohol]-mediated iROP of TMC(OMe)2: synthesis of P(TMC(OMe)2). | ||
Experimental section
Materials
All manipulations involving air-sensitive compounds (that is those carried out with the zinc complex) were performed under inert atmosphere (argon, <3 ppm of O2) using standard Schlenk, vacuum line and glovebox techniques. Solvents were thoroughly dried and deoxygenated by standard methods and distilled before use. CDCl3 was dried over a mixture of 3 and 4 Å molecular sieves. Benzyl alcohol (Acros) was distilled over Mg turnings under argon atmosphere and kept over activated 4 Å molecular sieves. 1,3-Propanediol (Acros) was dried over activated 4 Å molecular sieves. 2,2-Dimethoxypropylene carbonate (TMC(OMe)2) was prepared according to literature methods8a,b and isolated in 53% yield after 3 recrystallizations in diethyl ether [1H and 13C NMR: δOCH3 3.34 ppm, 49.7 ppm; δCH2OC(O)O 4.30 ppm, 70.2 ppm, Fig. S1 and S2†)]. [(BDIiPr)Zn(N(SiMe3)2)] was synthesized following the literature procedure.10 All other reagents were used as received (Aldrich).Instrumentation and measurements
1H (300 and 200 MHz) and 13C (75 and 50 MHz) NMR spectra were recorded in CDCl3 on Bruker Avance AM 300 and DPX 200 spectrometers at 23 °C. Chemical shifts (δ) are reported in ppm and were referenced internally relative to tetramethylsilane (δ 0 ppm) using the residual 1H and 13C solvent resonance.Average molar mass (nSEC) and molar mass distribution (w/n) values were determined by SEC in THF at 30 °C (flow rate = 1.0 mL min−1) on a Polymer Laboratories PL50 apparatus equipped with a refractive index detector and a ResiPore 300 × 7.5 mm column. The polymer samples were dissolved in THF (2 mg mL−1). All elution curves were calibrated with polystyrene (PS) standards (nSEC values are uncorrected for a possible difference in hydrodynamic volume of the polycarbonatesvs. PS). The SEC traces of the polymers all exhibited a unimodal and symmetrical peak.
The molar mass values of short-chain H-[P(TMC(OMe)2)]-OBn samples were determined by 1H NMR analysis, from the relative intensity of the signals of either the methylene hydrogens (C(OMe2)CH2OC(O), δ 4.23 ppm) or the methyl hydrogens (C(OMe2)CH2OC(O), δ 3.28 ppm) of the P(TMC(OMe)2) main chain relative to the methylene hydrogens of the (CO)OCH2C6H5 (δ 5.19 ppm) or of the C(OMe2)CH2OH (δ 3.82 ppm) chain-end. The number-average molar mass values thus obtained by 1H NMR (nNMR) were in close agreement with the ones calculated, as reported in Table 1.
| Entry | Catalyst | Alcohol | [TMC(OMe)2]0/[catalyst]0/[alcohol]0 | Temp/°C | Reaction timea/min | Convb (%) |
ntheoc/g mol−1 |
nNMRd/g mol−1 |
nSECe/g mol−1 |
w/nf |
TOF g/h−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction times were not necessarily optimized. b Monomer conversion determined by 1H NMR spectroscopy. c Calculated from [TMC(OMe)2]0/[BnOH or PPD]0 × monomer conversion × MTMC(OMe)2 + MBnOH or MPPD with MTMC(OMe)2 = 162 g mol−1, MBnOH = 108 g mol−1, MPPD = 76 g mol−1. d Determined by 1H NMR taking into account the monomer conversion. e Determined by SECvs. polystyrene standards (uncorrected raw data). f Molar mass distribution calculated from SEC traces. g Non-optimized turnover frequency expressed in molTMC(OMe)2 molcatalyst h−1 and calculated over the whole polymerization time. | |||||||||||
| 1 | (BDI)Zn(NTMS2) | BnOH | 500:1:5 |
60 | 90 | 96 | 15660 |
nd | 12450 |
1.28 | 320 |
| 2 | (BDI)Zn(NTMS2) | BnOH | 500:1:10 |
60 | 60 | 45 | 3750 | 3570 | 3000 | 1.18 | 225 |
| 3 | (BDI)Zn(NTMS2) | BnOH | 500:1:1 |
90 | 120 | 92 | 74630 |
nd | 70200 |
1.79 | 230 |
| 4 | (BDI)Zn(NTMS2) | BnOH | 500:1:2 |
90 | 60 | 98 | 39800 |
nd | 38800 |
1.36 | 490 |
| 5 | (BDI)Zn(NTMS2) | BnOH | 500:1:5 |
90 | 60 | 93 | 15200 |
nd | 17000 |
1.25 | 465 |
| 6 | (BDI)Zn(NTMS2) | BnOH | 500:1:15 |
90 | 90 | 95 | 5240 | 6120 | 7100 | 1.22 | 317 |
| 7 | (BDI)Zn(NTMS2) | BnOH | 500:1:25 |
90 | 105 | 89 | 3000 | 2980 | 3130 | 1.16 | 254 |
| 8 | (BDI)Zn(NTMS2) | BnOH | 500:1:25 |
90 | 120 | 95 | 3190 | 3250 | 4500 | 1.12 | 237 |
| 9 | Al(OTf)3 | BnOH | 500:1:5 |
60 | 330 | 28 | 4650 | nd | nd | nd | 25 |
| 10 | Al(OTf)3 | BnOH | 500:1:5 |
90 | 330 | 96 | 15660 |
nd | 10600 |
1.18 | 87 |
| 11 | Al(OTf)3 | BnOH | 500:1:5 |
110 | 330 | 0 | — | — | — | — | — |
| 12 | DMAP | BnOH | 500:1:5 |
90 | 330 | 45 | 7400 | 6980 | 6800 | 1.23 | 41 |
| 13 | TBD | BnOH | 500:1:5 |
90 | 180 | 99 | 16150 |
nd | 14100 |
1.71 | 165 |
| 14 | BEMP | BnOH | 500:1:5 |
90 | 180 | 100 | 16300 |
nd | 14300 |
1.53 | 167 |
| 15 | (BDI)Zn(NTMS2) | PPD | 500:1:5 |
90 | 3 | 96 | 15630 |
nd | 13990 |
1.66 | 160 |
| 16 | (BDI)Zn(NTMS2) | PPD | 500:1:10 |
90 | 1.5 | 93 | 7610 | 5800 | 6500 | 1.61 | 310 |
| 17 (ref. 5e) | BEMP | PPD | 500:1:5 |
90 | 180 | 100 | 16280 |
nd | 14300 |
1.53 | 167 |
Monomer conversions were calculated from 1H NMR spectra of the crude reaction mixtures, from the integration (Int.) ratio Int.P(TMC(OMe)2)/[Int.P(TMC(OMe)2) + Int.TMC(OMe)2], using either the methylene hydrogens (C(OMe2)CH2OC(O)) or the methyl hydrogens of the dimethyl acetal of P(TMC(OMe)2) at δ 4.23 ppm and 3.28 ppm, and of TMC(OMe)2 at δ 4.30 and 3.34 ppm, respectively.
MALDI-ToF
mass spectra were recorded with an AutoFlex LT high-resolution spectrometer (Bruker) equipped with a pulsed N2 laser source (337 nm, 4 ns pulse width) and a time-delayed extracted ion source. Spectra were recorded in the positive-ion mode using the reflectron mode and an accelerating voltage of 20 kV. The polymer sample was dissolved in THF (HPLC grade, 10 mg mL−1) and a solution (2:1 v/v) of α-cyano-4-hydroxycinnamic acid (10 mg mL−1) in acetonitrile (HPLC grade)/0.1% TFA was prepared. Both solutions were then mixed in a 1:1 volume ratio respectively, deposited sequentially on the sample target and then air-dried. Bruker Care Peptide Calibration and Protein Calibration 1 Standards were used for external calibration.
Differential scanning calorimetry (DSC) analyses were performed on a Setaram DSC 131 apparatus calibrated with indium at a rate of 10 °C min−1, under continuous flow of helium (25 mL min−1), using aluminium capsules (typically 10 mg of polymer). The thermograms were recorded according to the following cycles: −40 °C to +180 °C at 10 °C min−1; +180 °C to −40 °C at 10 °C min−1. Thermal gravimetric analyses were performed on a TA Instruments SDT 2960.
Mechanical properties of the polymers were evaluated at Total Petrochemicals Research Center Feluy, Belgium, using compression molded sheets. These H-shaped samples (length × width × thickness = 17 × 4 × 0.05 mm) were prepared from a mini max molder of Custom Scientific Instruments Inc at 160 °C for P(TMC(OMe)2), 220 °C for PTMC, using typically 100 mg of the polymer. Tensile tests were then carried out on seven samples at room temperature according to ASTMD 882 using a ZWICK (MEC 125/2) apparatus with a load cell 200 N at a cross-head speed of 10 mm min−1. Strength and elongation at break values were calculated from the dynamic tensile diagrams. The sample specimen deformation was measured from the grip-to-grip separation, which initially was 10 mm.
Typical polymerization procedure
In a typical experiment (Table 1, entry 6), [(BDIiPr)Zn(N(SiMe3)2)] (5.6 mg, 8.6 μmol), a solution of BnOH (5 equiv. vs.Zn, 4.7 μL, 43 μmol) in toluene (0.1 mL) and TMC(OMe)2 (500 equiv. vs. Zn, 0.500 g, 4.3 mmol) were charged in a Schlenk flask in the glovebox. The flask was then immersed in an oil bath preset at the desired temperature (60–90 °C) and the reaction mixture was stirred over the appropriate time (note that reaction times were not systematically optimized). The reaction was quenched by adding an excess of an acetic acid solution (ca. 1 mL of a 16.5 mmol L−1 solution in toluene). The resulting mixture was concentrated under vacuum and the conversion determined by 1H NMR analysis of the residue. The crude polymer was then dissolved in CH2Cl2 and purified upon precipitation in cold methanol, filtered and dried under vacuum. The final polymer was then analyzed by NMR and SEC, DSC and TGA. Noteworthy, the polycarbonates' purification upon its selective precipitation in MeOH is a procedure that simultaneously allows elimination of catalytic residues within the filtrate. Thus, taken into account that the iROP procedure typically involves very low initial catalytic loading,3 the polymers are thereby recovered with only minor traces—if any—of catalytic residues.Results and discussion
Binary catalytic systems that proved effective in the (i)ROP of the related unsubstituted TMC5 were evaluated in the (i)ROP of TMC(OMe)2 towards the synthesis of the corresponding homopolycarbonates P(TMC(OMe)2)s. Thus the highly active β-diiminate discrete zinc complex, [(BDIiPr)Zn(N(SiMe3)2)] (BDIiPr = 2-((2,6-diisopropylphenyl)amido)-4-((2,6-diisopropylphenyl)-imino)-2-pentene), the Lewis acidic triflate salt Al(OTf)3, and the organic bases, 4-N,N-dimethylaminopyridine (DMAP), 1.5.7-triazabicyclo-[4.4.0]dec-5-ene (guanidine, TBD) and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (phosphazene, BEMP), were successfully combined with a monoalcohol (benzyl alcohol, BnOH) or a diol (1,3-propanediol, PPD). The P(TMC(OMe)2)s thus synthesized displayed various molar masses ranging from 3000 to 70200 g mol−1 and showed a quite good degree of control over the molecular features. Selected results at a typical catalyst loading of 0.2 mol% ([TMC(OMe)2]0/[catalyst]0 = 500:1) are reported in Table 1.
Although the bulk11iROP of TMC(OMe)2 was successfully carried out at 60 °C, raising the polymerization temperature to 90 °C significantly improved the activity of both metallic catalytic systems, [(BDIiPr)Zn(N(SiMe3)2)]/BnOH and Al(OTf)3/BnOH (Table 1, entries 1 vs. 5, 9 vs. 10). At this latter temperature, the molar mass values measured by NMR and SEC were in good agreement with the ones expected (as determined from the monomer-to-alcohol ratio and the conversion), whereas the molar mass distribution values generally remained quite narrow. An increase in the relative amount of alcohol/chain transfer agent led to proportionally lower molar mass polycarbonates, as depicted in Fig. 1, in agreement with a controlled iROP process.3 This adequate compromised reaction temperature of 90 °C is higher than the one suitable for the iROP of TMC with this system under similar operating conditions. Indeed, at 60 °C, at [monomer]0/[(BDIiPr)Zn(N(SiMe3)2)]0/[BnOH]0 = 500:1:5, complete TMC conversion was reached within 7 min (TOFTMC = 4240 h−1),3,5a,b whereas 90 min were required to achieve almost quantitative TMC(OMe)2 conversion (TOFTMC(OMe)2 = 320 h−1; Table 1, entry 1).
![Dependence of the molar mass M̄n of P(TMC(OMe)2)s synthesized at 90 °C on the benzyl alcohol content at [TMC(OMe)2]0/[(BDI)Zn[N(SiMe3)2]]0 of 500 : 1. △, Experimental values determined by SEC; ■ theoretical values.](/image/article/2011/PY/c1py00405k/c1py00405k-f1.gif) | ||
| Fig. 1 Dependence of the molar mass n of P(TMC(OMe)2)s synthesized at 90 °C on the benzyl alcohol content at [TMC(OMe)2]0/[(BDI)Zn[N(SiMe3)2]]0 of 500:1. △, Experimental values determined by SEC; ■ theoretical values. | ||
Similarly, with the aluminium-based catalyst system, better activities were (more easily) obtained in the iROP of TMC than in that of TMC(OMe)2. At [monomer]0/[Al(OTf)3]0/[BnOH]0 = 500:1:5, 480 TMC equiv. were polymerized at 110 °C (TOFTMC = 480 h−1)3,5d while in the iROP of TMC(OMe)2, the highest—yet moderate—activity was observed at 90 °C (TOFTMC(OMe)2 = 87 h−1; Table 1, entry 10). Raising the temperature to 110 °C in the ROP of TMC(OMe)2 with this catalyst system led to degradation of the polycarbonate upon cleavage of the acetal group (Table 1, entry 11), whereas this same aluminium catalytic system was revealed to be most effective at 150 °C in the iROP of unsubstituted TMC (TOFTMC = 5880 h−1).3,5d
The decreased polymerization activities observed with TMC(OMe)2 as compared to TMC and metal-based catalysts most likely result from unfavorable interactions of the additional methoxy functionalities with the Lewis acidic metal centers, which compete with the effective activation of the carbonate moiety.
DMAP, TBD and BEMP, in association with BnOH at 90 °C, all ring-opened TMC(OMe)2 with the guanidine (TOFTMC(OMe)2 = 165 h−1; Table 1, entry 13) and phosphazene (TOFTMC(OMe)2 = 167 h−1; Table 1, entry 14) being both more active than the amine (TOFTMC(OMe)2 = 41 h−1; Table 1, entry 12). The organic bases were then revealed to be significantly more effective than the aluminium-based system (TOFTMC(OMe)2 = 87 h−1; Table 1, entry 10), but still less active than the zinc-based system (TOFTMC(OMe)2 = 465 h−1; Table 1, entry 5).12
Replacing benzyl alcohol with 1,3-propanediol in the iROP of TMC(OMe)2 promoted by [(BDIiPr)Zn(N(SiMe3)2)] or BEMP afforded the corresponding α-ω-dihydroxy telechelic polycarbonates, H-P(TMC(OMe)2)-H. Similar to the data obtained with BnOH, good activities and control in terms of molar mass and molar mass distribution of P(TMC(OMe)2) were reached with the diol, as previously observed in the corresponding PTMC syntheses5c,f (Table 1, entries 15–17).
1H NMR analyses of the precipitated polycarbonates prepared from these binary catalyst systems showed the main polymer chain typical resonances (δ 4.23, 3.28 ppm; CH2C(OMe)2CH2OC(O)O), indicating that no side processes (e.g., hydrolysis of the 2,2-dimethyl acetal functionalities) occurred, despite the use of relatively large amounts of alcohol (diol) and the presence of Lewis acidic metal centers (Fig. 2 and ESI†). Besides, low intensity resonances for both a hydroxymethyl (δ 3.82 ppm; CH2OH) and a benzyloxy carbonate group (δ 7.38, 5.19 ppm; PhCH2OC(O)O) were observed, in agreement with the expected formation of H-P(TMC(OMe)2)-OBn (Scheme 2).5e When PPD was used, the unique polymer chain-end identified by the characteristic triplet at δ 3.76 ppm representative of a hydroxymethyl end-group, supported the formation of α,ω-dihydroxy telechelic H-P(TMC(OMe)2)-H (Scheme 2, Fig. 2 and ESI†). No ether linkages (δ1H 3.4–3.1 ppm; δ13C 66.5–67.7 ppm) were detected in any spectrum of the polymers, thereby highlighting the ability of all initiating systems to prevent decarboxylation. The molar mass values of short-chain P(TMC(OMe)2)s, as determined by 1H NMR spectroscopy, were in close agreement with the ones calculated ntheo and the one measured by SEC, nSEC as well (Table 1).
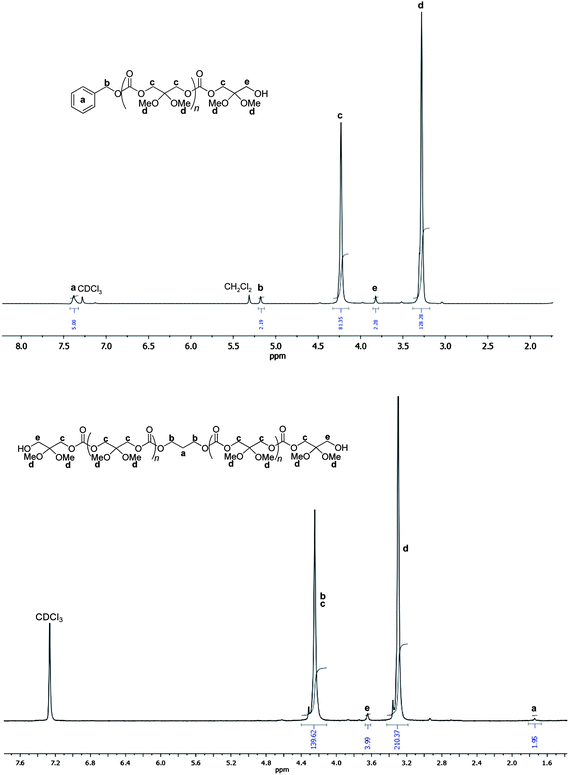 | ||
| Fig. 2 1H NMR (300 MHz, CDCl3, 23 °C) spectra of a H-P(TMC(OMe)2)-OBn (top) and H-P(TMC(OMe)2)-H (bottom) (Table 1, entries 2 and 16). | ||
These results were confirmed by MALDI-ToF mass spectrometry analyses of low molar mass P(TMC(OMe)2) samples. Analyses of a H-P(TMC(OMe)2)-OBn sample prepared from the BEMP/BnOH catalytic system (Table 1, entry 7) featured a major distribution of peaks unambiguously assignable to H-P(TMC(OMe)2)-OBn molecules cationized by Na+ ions (H-P(TMC(OMe)2)–OCH2Ph·Na+; main peak centered at m/z: 3209.5 g mol−1) with a repeat unit of 162.2 g·mol−1 (i.e., the molar mass of TMC(OMe)2; Fig. S3†).5e This clearly demonstrated that BnOH indeed acted as an effective co-initiator and a chain transfer agent in the iROP of TMC(OMe)2, ultimately end-capping the polycarbonate.
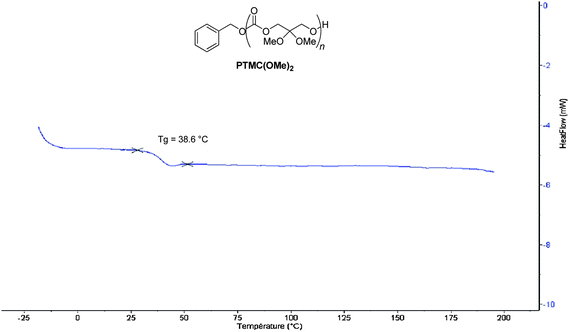
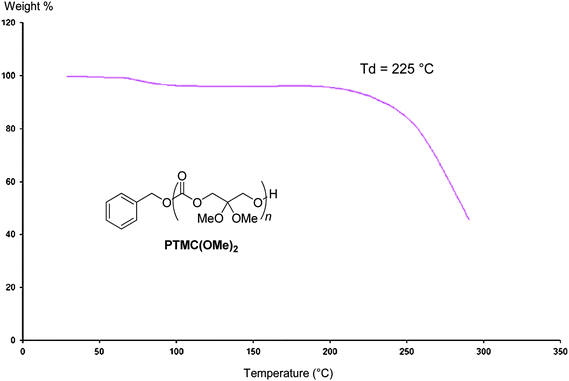
The thermal properties of P(TMC(OMe)2) samples were next investigated by DSC and TGA (see also Table S1 in the ESI†). The DSC trace of the polymer (Fig. 3) featured a relatively high temperature glass transition at Tg = 39 °C, in comparison to the negative Tg value (−15 °C; n = 10000 g mol−1)13 of TMC. The absence of a melting temperature transition highlighted the amorphous character of these functional polycarbonates similarly to PTMCs. The degradation temperature of PTMC(OMe)2, Td = 225 °C, measured by TGA (Fig. 4) is slightly lower than that of PTMC (Td = 230 °C).14
Mechanical properties of the P(TMC(OMe)2)s, especially the elastic modulus (E), the strain at break (σr) and the elongation at break (εr), were determined from tensile tests and compared to those of PTMC and poly(L-lactide). Illustrative values of each parameter are reported in Table 2 for all three polymers. PTMC exhibits both a very small elasticity modulus (E = 4.5 ± 1 MPa) and a quite high elongation at break (εr = 650 ± 77%) value, thus demonstrating its well known elastomeric characteristics (Table 2, entry 1). On the contrary, samples of PTMC(OMe)2 displayed a high E (3190 ± 70 MPa) value along with a low εr (9 ± 1%) (Table 2, entry 2), characteristic of a rigid material much like poly(L-lactide) (Table 2, entry 3). This was rather unexpected considering the amorphous nature of this polymer.
| Injection conditions | Tensile parameters | |||||
|---|---|---|---|---|---|---|
| Entry |
Polymer (n/g mol−1) |
Polymer temp/°C | Molding temp/°C | E a/MPa | σ r b/MPa | ε r c (%) |
| a Elastic modulus. b Strain at break. c Elongation at break. | ||||||
| 1 |
PTMC (95000) |
220 | 23 | 4.5 ± 1 | 1.8 ± 0.7 | 650 ± 77 |
| 2 |
PTMC(OMe)2 (70200) |
160 | 23 | 3190 ± 70 | 126 ± 3 | 9 ± 1 |
| 3 |
PLLA
3 (95000) |
190 | 23 | 3427 ± 71 | 140 ± 7 | 6 ± 1 |
Conclusions
Well-defined homopolymers derived from 2,2-dimethoxytrimethylene carbonate TMC(OMe)2 have been prepared with controlled molecular features by immortal ROP, under mild conditions (bulk, 60–90 °C), from various binary (metallo)organic catalysts/alcohol systems: H-P(TMC(OMe)2)-OBn and α,ω-dihydroxy telechelic H-P(TMC(OMe)2)-H of various molar masses ranging from 3000 to 70200 g mol−1 and quite narrow molar mass distribution values (w/n ≤ 1.79) were thus synthesized. These acetal-functionalized homopolycarbonates were all characterized at the molecular (NMR, SEC, MALDI-ToF-MS), thermal (DSC, TGA) and mechanical levels. Unlike PTMC which is elastomeric, mechanical properties of amorphous P(TMC(OMe)2) evidenced a rigid and brittle polymer material (E = 3190 ± 70 MPa, εr = 9 ± 1%), much like semi-crystalline poly(L-lactide).
Acknowledgements
Financial support of this research (PhD grant to M. H.) from Total Petrochemicals Co. is gratefully acknowledged. S.M. G gratefully acknowledges the Region Bretagne ACOMB research program and Rennes Métropole for equipment support.References and notes
- (a) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS; (b) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS; (c) T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS; (d) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS; (e) B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 11(49), 832–864 CrossRef.
- (a) F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Polym. Chem., 2011, 2, 528–533 RSC; (b) R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier and Y. Sarazin, Dalton Trans., 2010, 39, 8363–8376 RSC.
- (a) P. Brignou, M. Priebe Gil, O. Casagrande, Jr, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS; (b) P. Brignou, M. J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2011, 44, 5127–5135 CrossRef CAS.
- (a) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem.–Eur. J., 2008, 14, 8772–8775 CrossRef CAS; (b) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Adv. Synth. Catal., 2009, 351, 1312–1324 CrossRef CAS; (c) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2009, 30, 2128–2135 CrossRef CAS; (d) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, ChemCatChem, 2010, 2, 306–313 CrossRef CAS; (e) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem.–Eur. J., 2010, 16, 13805–13813 CrossRef CAS; (f) M. Helou, J.-F. Carpentier and S. M. Guillaume, Green Chem., 2011, 13, 266–271 RSC.
- (a) C. Guillaume, J.-F. Carpentier and S. M. Guillaume, Polymer, 2009, 50, 5909–5917 CrossRef CAS; (b) C. Guillaume, N. Ajellal, J.-F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 907–917 CrossRef CAS.
- A. N. Zelikin and D. Putnam, Macromolecules, 2005, 38, 5532–5537 CrossRef CAS.
- (a) A. N. Zelikin, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2006, 7, 3239–3244 CrossRef CAS; (b) P. N. Zawaneh, A. M. Doody, A. N. Zelikin and D. Putnam, Biomacromolecules, 2006, 7, 3245–3251 CrossRef CAS; (c) J. R. Weiser, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2011, 4, 977–986 Search PubMed.
- For a brief mention of the synthesis of P(TMC(OMe)2) using a phosphazene-type catalyst, see ref. 5e.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS; (b) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS.
- Bulk operating conditions were preferentially selected to remain closer to environmentally friendly procedures as defined by one of the principles of green chemistry in Green Chemistry: Theory and Practice, ed. P. T. Anastas and J. C. Warner, Oxford University Press, Oxford, 1998 Search PubMed.
- Note that this general trend only applies in the case of purified monomers. Indeed, upon polymerizing technical grade TMC, the organic- and aluminium triflate/BnOH systems revealed to be more robust and more efficient than systems based on [(BDIiPr)Zn(N(SiMe3)2)].
- (a) I. Palard, M. Schappacher, B. Belloncle, A. Soum and S. M. Guillaume, Chem.–Eur. J., 2007, 13, 1511–1521 Search PubMed; (b) K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736–1740 Search PubMed.
- F. Nedeberg, V. Trang, R. C. Pratt, A. F. Mason, C. W. Frank, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 3294–3297 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR and MALDI-ToF mass spectra and thermal properties of the monomer and polymers. See DOI: 10.1039/c1py00405k |
| This journal is © The Royal Society of Chemistry 2011 |