Aliphatic polycarbonates and poly(ester carbonate)s from fatty acid derived monomers†
Arvind S.
More
ab,
Dnyaneshwar V.
Palaskar
ab,
Eric
Cloutet
ab,
Benoit
Gadenne
c,
Carine
Alfos
c and
Henri
Cramail
*ab
aUniversité de Bordeaux, Laboratoire de Chimie des Polyméres Organiques, UMR 5629, IPB/ENSCBP, 16 avenue Pey-Berland, F-33607, Pessac Cedex, France
bCentre National de la Recherche Scientifique, Laboratoire de Chimie des Polyméres Organiques, UMR 5629, F-33607, Pessac Cedex, France. E-mail: cramail@enscbp.fr
cITERG, 11 rue Gaspard Monge, Parc Industriel, Pessac Cedex, F-33600, France
First published on 10th October 2011
Abstract
Fatty acid derivatives were efficiently used as starting materials for the synthesis of polycarbonates and poly(ester carbonate)s. A novel AB-type self-condensable monomer, ethyl(9-hydroxy-10-methoxyoctadecyl)carbonate (EHMOC) and a dicarbonate monomer, 4-[(ethoxycarbonyl)oxy]butyl-12-[(ethoxycarbonyl)oxy]octadec-9-enoate (EOBEOE) were prepared from oleyl alcohol and ricinoleic acid, respectively. Of these, EHMOC was polymerized by the alcohol–carbonate exchange self-polycondensation approach, while EOBEOE was polycondensed with various biobased diols to give polycarbonates and poly(ester carbonate)s, respectively. The monomers and polymers were well characterized by FTIR and 1H-NMR spectroscopy. The 13C-NMR spectroscopy revealed the formation of randomly distributed sequences in the poly(ester carbonate)s due to the carbonate interchange reaction. An unexpected formation of polyricinoleate was observed and confirmed by NMR and MALDI-TOF spectroscopy. Most of the polymers displayed good thermal stability with the temperature at 10% weight loss in the range 273–325 °C. Due to the presence of aliphatic segments, these materials exhibit very low glass transition temperature.
Introduction
Presently, most of the industrial chemicals are manufactured from petroleum resources. However, rapid depletion and price increase of fossil resources are encouraging chemists to orient their research towards designing chemicals and materials from renewable feedstocks, with an increased awareness of environmental and industrial impact. Given the availability, sustainability and biodegradability of natural resources like vegetable oils and natural fats, rigorous initiatives have been recently undertaken, particularly in the field of polymer science and technology, to prepare polymeric materials based on renewable resources.1–4Based on the fatty acid derivatives (FAD), a lot of work has been done so far in the field of polyurethanes,2,5–7 polyesters,8–11polyamides,4,12,13etc. However, not as much attention has been paid in the preparation of polycarbonates starting from FAD. Aliphatic polycarbonates and poly(ester carbonate)s are a class of biocompatible polymers that have recently received considerable attention due to their potential applications in the medical field.14,15 Furthermore, they find applications as oligomer and polymer additives with plasticizing or thickening properties.16,17 While avoiding the use of toxic phosgene, there are several other routes employed for the synthesis of polycarbonates, such as the copolymerization of epoxides with carbon dioxide in the presence of organometallic catalyst,18,19 the direct condensation of diols with carbon dioxide or alkali metal carbonates,20,21 the ring opening polymerization of cyclic carbonate monomers,22–25 the enzyme catalyzed synthesis of polycarbonates14,26 and the carbonate interchange reaction between diols and carbonates.27 The use of carbon dioxide has appeared to be a very promising means to polycarbonates.4,28 There have been several reports on the utilization of carbon dioxide as feedstock for synthesis of polycarbonates.4,18,28,29 Recently, Hu et al. reported the copolymerization of furfuryl glycidyl ether and carbon dioxide using a rare earth ternary catalyst.18 Enzymatic synthesis of poly(ester carbonate)s with higher molar mass has been described by Zini et al.14 Brignou et al. utilized green renewable acids to derive polycarbonatesvia their cyclization to the cyclic carbonates followed by ring opening polymerization.22
In spite of the accumulation of a considerable amount of literature on polycarbonates from renewable resources, very little data are available on the direct use of fatty acid derivatives as monomers.30,31 The inherent presence of functionalities (hydroxyl functions, double bonds) and the ability to undergo various organic transformations make fatty acids desirable precursors. In the last few years, our research group has been focusing on the development of new polymers from vegetable oils.32–34 Recently, we have demonstrated the use of sunflower oil- and castor oil-based monomers for the synthesis of polyurethanes.32,33 In line with these studies, we report in this manuscript the synthesis of a new linear dicarbonate monomer and an AB-type self-condensable monomer starting from fatty acid derivatives and their utilization for the synthesis of poly(ester carbonate)s (PEC) and polycarbonate (PC) respectively, viacarbonate interchange reaction.
Experimental section
Materials and instrumentation
4-Hydroxybutyl-12-hydroxyoctadec-9-enoate (HBHOE) was provided by ITERG Pessac, France and used as received. Oleyl alcohol (85% technical grade), meta-chloroperbenzoic acid (mCPBA), titanium tetrabutoxide [Ti(n-BuO)4], diethyl carbonate (DEC), dibutyltin oxide, Amberlyst 15, 1,6-hexanediol, polycaprolactone diol (Mn 1250) (PCL1250), and poly(ethylene glycol) (Mn 200) (PEG200) were purchased from Sigma-Aldrich and used as received. Isosorbide was purchased from Alfa-Aesar and used as received. Poly(propylene glycol) (Mn 400) (PPG400) was received from Dow and used as received. The solvents were of reagent grade quality and were used as received.
1H and 13C NMR spectra were recorded using a Bruker AC-400 NMR at room temperature by dissolving the samples in CDCl3. Size exclusion chromatography (SEC) analyses were performed at room temperature in THF with a setup consisting of a WATERS 880-PU pump and a series of three microstyragel columns (pore size 103, 105 and 106 Å). The elution of the filtered samples was monitored using simultaneous UV and refractive index detections. The elution times were converted to molar mass using a calibration curve based on low dispersity (Mw/Mn) polystyrene (PS) standards. Infrared spectra were obtained on a Bruker-Tensor 27 spectrometer using the attenuated total reflection (ATR) mode. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed by the CESAMO (Bordeaux, France) on a Voyager mass spectrometer (Applied Biosystems). The instrument is equipped with a pulsed nitrogen laser (337 nm) and a time-delayed extracted ion source. Spectra were recorded in the positive-ion mode using the reflectron and with an accelerating voltage of 20 kV. Polymer samples were dissolved in THF at 10 mg mL−1. The dithranol matrix solution was prepared by dissolving 10 mg in 1 mL of dichloromethane. A methanol solution of cationization agent (NaI, 10 mg mL−1) was also prepared. The solutions were combined in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 volume ratio of matrix to polymer to cationization agent. Several microlitres of the obtained solution were deposited onto the sample holder and vacuum dried. Differential scanning calorimetry (DSC) thermograms were measured using a DSC Q100 apparatus from TA Instruments. Polymer samples were first heated from −100 to 150 °C and then the glass transition temperatures were calculated from a second heating run. All runs were performed at a rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TGA-7 system at a heating rate of 15 °C minute−1 under nitrogen atmosphere.
:1:1 volume ratio of matrix to polymer to cationization agent. Several microlitres of the obtained solution were deposited onto the sample holder and vacuum dried. Differential scanning calorimetry (DSC) thermograms were measured using a DSC Q100 apparatus from TA Instruments. Polymer samples were first heated from −100 to 150 °C and then the glass transition temperatures were calculated from a second heating run. All runs were performed at a rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TGA-7 system at a heating rate of 15 °C minute−1 under nitrogen atmosphere.
(10.89 g, yield: 86%). IR (cm−1): 1740 (–OCOO). 1H-NMR (400 MHz, CDCl3): 0.80 (3H, t, J = 6.9 Hz, CH3), 1.10–1.40 (methylene protons and CH2–OCOO–CH2CH3), 1.59 (2H, q, J = 6.3 Hz, CH2–CH2OCOO), 1.92 (4H, m, J = 6.6 Hz, CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2), 4.04 (2H, t, J = 6.4 Hz, CH2–OCOO–CH2CH3), 4.11 (2H, q, J = 7.1 Hz, CH2–OCOO–CH2CH3), 5.27 (2H, m, J = 5.5 Hz, CHCH).
CH–CH2), 4.04 (2H, t, J = 6.4 Hz, CH2–OCOO–CH2CH3), 4.11 (2H, q, J = 7.1 Hz, CH2–OCOO–CH2CH3), 5.27 (2H, m, J = 5.5 Hz, CHCH).
(b) Oleyl carbonate (2: 3 g, 8.82 mmol) and mCPBA (2.28 g, 13.23 mmol) were dissolved in DCM (50 mL) and the reaction mixture was stirred at room temperature for 5 h. The reaction mixture was filtered, and the solution was washed with aqueous sodium bicarbonate (3 × 25 mL), followed by water (3 × 25 mL). The organic layer was separated and dried over anhydrous sodium sulfate. The solvent was removed by a rotary evaporator to obtain intermediate 3.
(2.82 g, yield: 90%). IR (cm−1): 1741 (–OCOO), 841 cm−1 (epoxy). 1H-NMR (400 MHz, CDCl3): 0.81 (3H, t, J = 6.6 Hz, CH3), 1.10–1.50 (methylene protons, CH2–OCOO–CH2CH3 and CH2–epoxide–CH2), 1.58 (2H, m, J = 6.5 Hz, CH2–CH2OCOO), 2.57 (m, J = 4.9 Hz, trans epoxy protons), 2.83 (m, J = 5.5 Hz, cis epoxy protons), 4.05 (2H, t, J = 6.8 Hz, CH2–OCOO–CH2CH3), 4.12 (2H, q, J = 7.1 Hz, –OCOO–CH2CH3).
(c) Epoxidised oleyl carbonate (3: 2 g, 5.61 mmol), Amberlyst 15 (0.02 g) and excess methanol (50 mL) were refluxed for 12 h. The reaction mixture was filtered and methanol was removed using a rotary evaporator. The reaction mixture was dissolved in DCM (100 mL) and washed with water (3 × 50 mL). The DCM layer was dried over anhydrous sodium sulfate, filtered and the solvent was removed to obtain 4/4′.
(1.90 g, yield: 87%). IR (cm−1): 3450 (OH), 1740 (–OCOO). 1H-NMR (400 MHz, CDCl3): 0.81 (3H, t, J = 6.2 Hz, CH3), 1.10–1.70 (methylene protons, CH2–OCOO–CH2CH3), 2.24 (J = 4.1 Hz, CH–OH), 2.91 (1H, m, J = 6.2 Hz, CH–OCH3), 3.40 (1H, m, J = 4.1 Hz, CH–OH), 3.41 (3H, s, –OCH3), 4.05 (2H, t, J = 6.9 Hz, CH2–OCOO–CH2CH3), 4.13 (2H, q, J = 7.1 Hz, –OCOO–CH2CH3). 13C-NMR (400 MHz, CDCl3): 154.78, 83.73, 72.13, 67.31, 63.20, 57.49, 33.06, 32.85, 29.28, 28.56, 27.59, 25.17, 23.12, 22.14, 13.39.
(6.26 g, yield: 90%). IR (cm−1): 1742 (–OCOO). 1H-NMR (400 MHz, CDCl3): 0.81 (3H, t, J = 7.1 Hz, CH3), 1.10–1.80 (methylene protons and –OCOO–CH2CH3), 1.93 (2H, m, J = 6.9 Hz, CHCH–CH2), 2.10–2.40 (4H, m, J = 9.1 Hz, CH2–COO and CH2–CHCH), 3.90–4.20 (8H, m, COO–CH2–(CH2)2–CH2–OCOO–CH2 and CH–OCOO–CH2), 4.62, 4.80 (m, J = 6.16 and 6.8 Hz, CH–OCOO–CH2), 5.20–5.50 (2H, m, CHCH). 13C-NMR (400 MHz, CDCl3): 173.93, 155.20, 150.08, 133.03, 123.92, 78.27, 77.29, 67.50, 67.26, 63.89, 60.54, 35.03, 34.01, 31.69, 29.37, 27.57, 25.76, 22.67, 14.17.
IR (cm−1): 1741 (–OCOO). 1H-NMR (400 MHz, CDCl3): 0.79, 1.00–1.80, 3.10, 3.22, 3.31, 4.10, 4.67.
Results and discussion
The abundant availability and ability to undergo various organic transformations have made plant oils an important source for preparation of monomers.35 Among these, oleic and ricinoleic acids are extensively used. The former is the most common mono-ene, found in most plant and animal lipids, the major fatty acid in olive oil and several nut oils, while the latter is a fatty acid bearing a hydroxyl moiety and the major constituent in castor oil. Oleyl alcohol can be obtained either by reduction of ester of oleic acid or can be isolated from inedible beef fat.36,37 In the present study we have utilized oleyl alcohol and ricinoleic acid as precursors for the synthesis of polycarbonate and poly(ester carbonate)s, respectively.
Scheme 1 illustrates the synthesis of EHMOC as an AB-monomer for the synthesis of polycarbonate starting from oleyl alcohol. First of all, oleyl carbonate (see Fig. S1 in the ESI†) (2) was prepared from technical grade oleyl alcohol (1) by the dialkyl carbonate interchange reaction. The reaction was carried out in excess of DEC using dibutyltin oxide as a catalyst. The epoxidation of the double bond in oleyl carbonate was carried out with mCPBA as the oxidizing agent in quantitative yield (see Fig. S2 in the ESI†). The acid catalyst Amberlyst 15 was used to open the epoxide with an excess of methanol leading to a mixture of two regio-isomers of EHMOC (4/4′), an AB-monomer. For the simplicity of the reaction schemes, we have written one isomer structure in Fig. 1. The structures of 4/4′ and the intermediates involved in the reaction were confirmed by FTIR and 1H- and 13C-NMR spectroscopy. The FTIR spectrum of 4/4′ showed a band at 3450 cm−1, corresponding to hydroxyl, and another band at 1740 cm−1, characteristic of CO of carbonate.
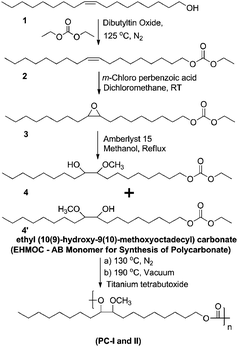 | ||
| Scheme 1 Synthesis of EHMOC (starting from oleyl alcohol) and corresponding polycarbonate. | ||
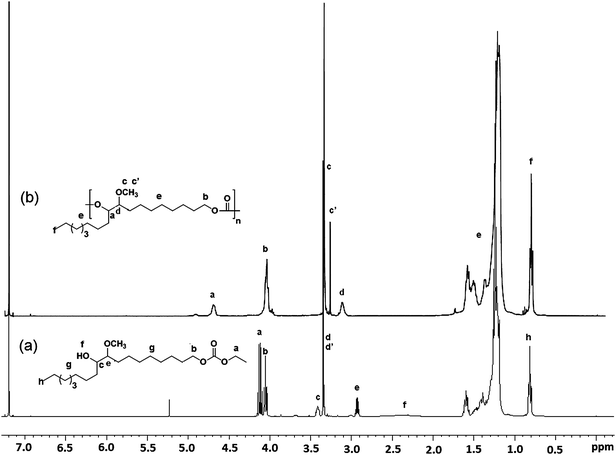 | ||
| Fig. 1 Stacked 1H-NMR spectra of (a) EHMOC and (b) polycarbonate (PC-I) derived by self-polycondensation of EHMOC. | ||
In the 1H-NMR spectrum of EHMOC, 4/4′ (Fig. 1a), a triplet (b) and a quartet (a) were observed in the region 4.00–4.2 δ ppm, corresponding to methylene protons adjacent to carbonate linkage. Two significant peaks at 3.40 and 2.91 δ ppm were observed due to the methine protons (c and e) adjacent to hydroxyl and methoxy moieties, respectively. The methoxy protons (d and d′) appeared as a singlet at 3.35 δ ppm. In the 13C-NMR spectrum of EHMOC, 4/4′, carbonyl carbon displayed a signal at 154.78 δ ppm. The carbons attached to the methoxy and hydroxyl groups appeared at 83.73 and 72.13 δ ppm, respectively (see Fig. S3 in the ESI†).
To synthesize the dicarbonate EOBEOE, 6, HBHOE (see Fig. S4 in the ESI†) was subjected to carbonate interchange reaction in excess of DEC in the presence of dibutyltin catalyst (Scheme 2). The dicarbonate precursor was obtained as viscous yellow oil. The FTIR spectrum of EOBEOE revealed an absorption band at 1742 cm−1 due to the presence of carbonate function which overlaps with the ester function band. The 1H-NMR spectrum of EOBEOE was provided in Fig. 2a along with the assignment of protons. The downfield shifting of both, the methylene proton signals (c) and methine proton (b) attached to the carbonate functions, confirmed the formation of dicarbonate moiety. The dicarbonate formation was also confirmed by 13C-NMR spectroscopy (see Fig. S5 in the ESI†). Carbons of the carbonate functions were observed as two different signals at 155.20 and 155.08 δ ppm. The ester carbon appeared most downfield at 173.93 δ ppm, while the olefinic carbons appeared at 133.03 and 123.92 δ ppm.
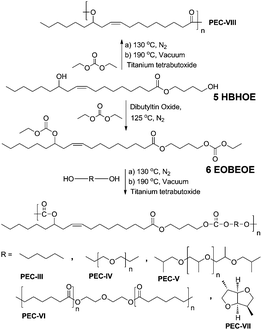 | ||
| Scheme 2 Synthesis of EOBEOE and poly(ester carbonate)s therefrom. | ||
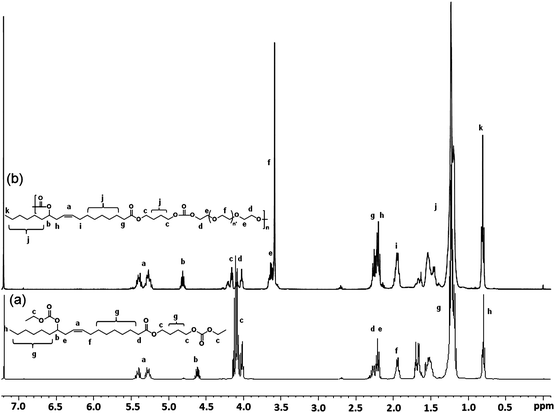 | ||
| Fig. 2 Stacked 1H-NMR spectra of (a) EOBEOE and (b) poly(ester carbonate) (PEC-IV) derived by polycondensation of EOBEOE and PEG200. | ||
Polymerization
The synthetic approach used in this work is the carbonate interchange reaction with an alcohol, using an organometallic catalyst. The polycondensation reaction was carried out in bulk and in two stages. In the first stage, reactants were heated at 130 °C under a slow stream of nitrogen to form oligomers. The formed oligomers were then reacted at 190 °C under reduced pressure so as to form higher molar mass polymers.The AB-monomer EHMOC (4/4′), bearing both hydroxyl and carbonate moieties, was subjected to self-polycondensation in bulk and in two stages using titanium tetrabutoxide as a catalyst (Scheme 1). The reaction proceeded in a homogeneous manner and a viscous polymer was obtained at the end of the reaction. The reaction conditions and results of polymerization are given in Table 1. It was found that when the second stage of polymerization was continued for extended hours, a significant increase in the molar mass was observed (see Table 1, entry PC-II). The polycarbonate was characterized by FTIR, 1H- and 13C-NMR spectroscopy (Fig. 1b and S6 in the ESI†). The band at 3450 cm−1 corresponding to the hydroxyl disappeared and the carbonyl band shifted to 1742 cm−1. In the 1H-NMR spectrum of polycarbonate, the methine proton (a), previously adjacent to the hydroxyl group, showed a downfield shift due to the newly formed carbonate linkage. The other methine proton (d) nearby to the methoxy moiety also shifted to the downfield at 3.10 δ ppm.
| Entry | Monomer | Diol | Timeb (a/b)/h | SEC c | Thermal data | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| M n/g mol−1 | M w/g mol−1 | M w/Mn | T g/°C | T 0 d/°C | T 10 e/°C | T 50 f/°C | ||||
| a Polycarbonate from self-polycondensation of AB-monomer. b Two stage polycondensation: First stage: 130 °C, N2; second stage: 190 °C, vacuum. c Based on PS calibration. d Initial decomposition temperature. e Temperature at which 10% weight loss is observed. f Temperature at which 50% weight loss is observed. | ||||||||||
| PC-I a | EHMOC 4,4′ | — | 5/5 | 4900 | 9200 | 1.8 | −58 | 193 | 273 | 339 |
| PC-IIa | EHMOC 4,4′ | — | 5/8 | 13600 |
24400 |
1.7 | −59 | 201 | 306 | 333 |
| PEC-III | EOBEOE 6 | Hexanediol | 5/8 | 10600 |
20400 |
1.9 | −68 | 191 | 300 | 355 |
| PEC-IV | EOBEOE 6 | PEG200 | 5/8 | 6200 | 9700 | 1.5 | −71 | 176 | 288 | 367 |
| PEC-V | EOBEOE 6 | PPG400 | 5/8 | 8700 | 14500 |
1.6 | −65 | 185 | 296 | 352 |
| PEC-VI | EOBEOE 6 | PCL1250 | 5/8 | 14700 |
26200 |
1.8 | −66 | 160 | 275 | 332 |
| PEC-VII | EOBEOE 6 | Isosorbide | 5/8 | 10800 |
19200 |
1.7 | −62 | 136 | 290 | 348 |
| PEC-VIII | DEC | HBHOE 5 | 5/8 | 4500 | 6700 | 1.5 | −75 | 226 | 325 | 351 |
The difunctional carbonate monomer, EOBEOE, 6, was used to synthesize a series of poly(ester carbonate)s (PEC) with varying diols (Scheme 2). The diols chosen for the polycondensation reaction could be obtainable from biomass. The reactions were carried out in bulk. The reactants remain homogeneous in the system and viscous PECs were obtained. The results of the polymerization are given in Table 1. SEC data indicate the formation of reasonably good molar mass polymers. However, the molar mass values provided by SEC should not be taken as absolute values as the SEC calibration was carried out using polystyrene standards (see SEC traces in Fig. S7 in the ESI†). The number average molar masses of the PECs were in the range 4500 to 14700 g mol−1 with dispersity (Mw/Mn) in the range 1.5–1.9.
The PECs obtained were characterized by FTIR and 1H- and 13C-NMR spectroscopy (see Fig. S8–13† for NMR spectra of PECs). A characteristic band of the carbonate moiety at around 1741 cm−1 was observed for all PECs.
A representative 1H-NMR spectrum of the PEC derived from EOBEOE with PEG200 along with assignments is shown in Fig. 2b. The ethylenic protons (a) appeared as a multiplet in the range 5.20–5.55 δ ppm. The methine proton (b) appeared as a multiplet in the region 4.70–4.90 δ ppm. The methylene protons (c and d), adjacent to the carbonate and ester bonds, split as a multiplet in the range 3.90–4.20 δ ppm. The methylene protons (e and f) of the PEG sequence away from the carbonate linkage appeared as a multiplet in the range 3.40–3.75 δ ppm. The methylene protons neighboring the ester linkage (g) and those nearby the double bond (h and i) displayed two multiplets at 1.85–2.90 δ ppm.
While studying 13C-NMR spectra of PECs, it is observed that during the course of polycondensation, the dicarbonate-EOBEOE and the respective diol could have been reacted with each other in various ways. The random sequences in the polycarbonate due to the asymmetric carbonate interchange reactions is well documented in the literature,38–41 particularly when the reactions were carried out in bulk. The possibility of asymmetric carbonate interchange reaction between EOBEOE and diols at high temperature, under the influence of a catalyst, may lead to the formation of different types of monomer unit sequences.
We attempted to characterize the monomer unit sequences in the polymer chains with 13C-NMR spectroscopy. In all 13C-NMR spectra, we observed more than one peak belonging to the carbons of ester and carbonate respectively at 173 and 155 δ ppm, suggesting the presence of several sequences per repeating unit in the polymer chain (Fig. 3).
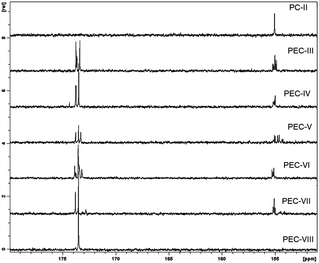
We tried to figure out the probable structures formed during the reaction (Fig. 4). We assumed that, besides the reaction of the dicarbonate monomer and the respective diol, five monomer units could have been formed during the polycondensation reaction, resulting in a plethora of random monomer sequences in the polymer chains. The dicarbonate-EOBEOE may undergo carbonate interchange reaction either fully or partially, giving rise to the parent diol—B3B3 (HBHOE) and two AB type monomers A2B2 and B1A1. In a similar way, the diol used may transform into A3A3 (dicarbonate) and B4A4 (AB type monomer) due to the probable exchange of carbonate. These structures suggest many ways in which they could arrange themselves in the polymer chains. Including the formation of the expected AA-BB sequence, there are several possibilities of monomer sequences in the polymer chain (Fig. 4). We observed another interesting behavior during a model polycondensation reaction of HBHOE and DEC (PEC-VIII). Indeed, the 13C-NMR spectroscopy only revealed, in the downfield region, a single peak at around 173 δ ppm corresponding to the ester carbon (Fig. 3 and S13 in the ESI†). No peak corresponding to the carbonate carbon at around 155 δ ppm in the 13C-NMR spectrum was observed (see Fig. S13 in the ESI†). Also in the 1H-NMR spectrum (see Fig. S13 in the ESI†), the methylene protons in butanediol adjacent to ester and hydroxyl group appeared as end functionalities at around 4.00 δ ppm, while the proton adjacent to the hydroxyl group has been shifted to the downfield. This suggests that during the first stage of oligomer formation, HBHOE did not react with DEC but the major reaction that could have taken place is polytransesterification. At higher temperature (190 °C), under vacuum, the unreacted DEC and butanediol could have been removed leading to the polyricinoleate. Indeed, the 1H-NMR spectrum agrees with the structure of polyricinoleate42 (see Fig. S13 in the ESI†). The formation of polyricinoleate was confirmed by MALDI-TOF analysis and it is discussed in the next section.
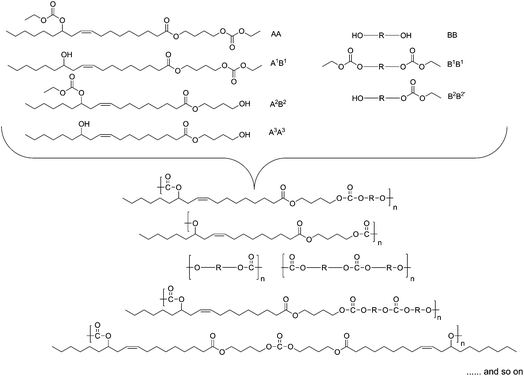 | ||
| Fig. 4 Possible monomer sequences in the repeat units of PECs. | ||
MALDI-TOF mass spectroscopy analysis
We attempted to analyze all the polymers by MALDI-TOF mass spectroscopy. The full desorption of polymers was found to be difficult, irrespective of the matrix used, for example, dithranol, trans-3-indoleacrylic acid, or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile. In addition, the spectra obtained were found to be complex, due to the several monomer sequences as well as the dispersity of the polymers.The spectra of poly(ester carbonate)s (Fig. S15–19 in the ESI†) were not very informative. However, the spectrum of polycarbonate (PC-II) was clearly defined (Fig. 5). The MALDI-TOF spectrum of PC-II was consistent with the expected structure; the peaks were characterized by the mass increment of 342 Da from one major peak to the next, and were equal to the mass of the repeating unit. A series of peaks at Mn 2807.6 Da corresponding to the structure [H + (342)n + (OCH2CH3) + Na+] was found. The most abundant series of peaks was corresponding to the Na adducts.
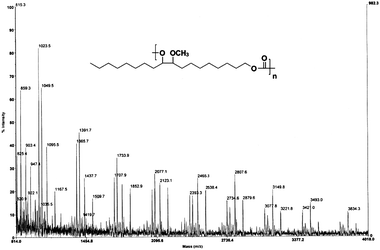 | ||
| Fig. 5 MALDI-TOF mass spectrum of polycarbonate (PC-II) derived by self-polycondensation of EHMOC. | ||
We have already seen that the reaction of HBHOE and DEC resulted in polyricinoleate (PEC-VIII). The formation of polyricinoleate was confirmed by MALDI-TOF spectroscopy. The spectrum was consistent with the polyricinoleate structure showing a major series of signals with an interval of 280, which corresponds to the repeat unit of polyricinoleate. We found peaks at m/z (1 + 280n + 89 + Na) = 1515.0 (n = 5), 1795.1 (n = 6), 2075.3 (n = 7), and so on corresponding to the polyricinoleate ended by two hydroxyl moieties (Fig. 6).
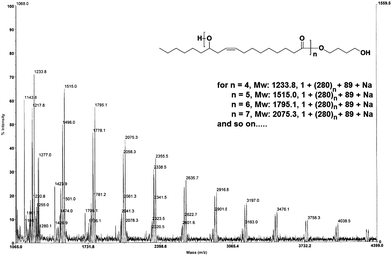 | ||
| Fig. 6 MALDI-TOF mass spectrum of polyricinoleate (PEC-VIII). | ||
Thermal characterization
The thermal behavior of the PC and PECs was evaluated by DSC and TGA. Tg values were obtained after second heating scans of polymer samples at a heating rate of 10 °C min−1 (Table 1). All polymers were amorphous in nature and hence a unique and well-identified glass transition is observed in all systems (see Fig. S20 in the ESI†). No melt endothermal behavior was detected in DSC. The PC derived from the AB-monomer displayed a Tg at −58 °C (see Table 1, entry PC-I). The glass transition temperatures for PECs derived from EOBEOE biscarbonate and various diols were in the range −62 to −71 °C, lower than the reported Tg values for poly(alkylene carbonate)s prepared from dimethyl carbonate and diols with 4 (−39.8 °C), 6 (−11.4 °C) and 8 (−51.7 °C) carbons, respectively.27 It is obvious that the aliphatic content, pendant alkyl chain and preformed ester linkage are responsible for the decrease in glass transition temperature. This is consistent with the generally accepted concept that the longer linear methylene units in a polymer main chain alter the macromolecular dynamics because of the increasing flexibility. The polymer (PEC-VIII) derived from HBHOE and DEC displayed the lowest glass transition temperature at −75 °C (see Table 1, entry PEC-VIII). The reported Tg for polyricinoleate is −74.8 °C42 and this supports our data that during the polycondensation reaction of HBHOE and DEC, instead of poly(ester carbonate), polyricinoleate has been formed. The incorporation of an isosorbide unit into the PEC was a thought to increase the Tg, however, the corresponding PEC displayed a Tg at −62 °C (see Table 1, entry PEC-VII). The possible reason for such a low Tg can be explained by the combined effect of the flexible alkyl spacers and the pendant alkyl chains that overpowered the rigidity of the isosorbide unit.The thermal stability of PC and PECs was studied by TGA in a nitrogen atmosphere and the obtained data are shown in Table 1 (see Fig. S21 in the ESI†). The T0 and T10 values are important criteria indicating the thermal stability of the polymers. Except PEC-VI and PEC-VII, all polymers exhibited T0 above 175 °C. The T10 and T50 values for polymers were in the range 273–325 °C and 332–367 °C, respectively. Lower thermal stability of the PC and PECs could be attributed to the thermally induced decarboxylation reactions.43 The early initial weight loss in isosorbide containing PEC (see Table 1, entry PEC-VII) was associated with the loss of absorbed water and is less than 0.5%. While comparing the thermal stability trend in PECs, we observed that PECs with PEG200 (see Table 1, entry PEC-IV) and PPG400 (see Table 1, entry PEC-V) as a comonomer displayed higher T10 value (288 and 296 °C) compared to the PEC containing PCL1250 (see Table 1, entry PEC-VI) (275 °C). This could be logically explained in terms of the higher thermal stability of the ether linkages over the ester ones. As mentioned earlier, the reaction of HBHOE and DEC (see Table 1, entry PEC-VIII) yielded polyricinoleate with a structure analogous to PC-I and II. It is worth noting that the thermal stability of the polymer derived from HBHOE with DEC (PEC-VIII) displayed higher thermal stability than PC (PC-I and II). This could be attributed to the fact that ester linkages (PEC-VIII, polyricinoleate) are less thermolabile in comparison to the carbonate moieties (PC-I and PC-II, polycarbonates). In spite of having aliphatic units in the main chain, the polymers exhibited fair thermal stability. A negligible weight residue at 700 °C for all polymers was observed, which showed complete decomposition of the polymer backbone.
Conclusion
In summary, we have synthesized a new (alcohol–carbonate) AB-self-condensable monomer and a dicarbonate monomer starting from fatty oleyl alcohol and ricinoleic acid, respectively using optimized organic transformations. The monomers were of polymerization grade and were thoroughly characterized by FTIR and 1H- and 13C-NMR spectroscopy. These monomers were utilized for the synthesis of a series of novel poly(ester carbonate)s and polycarbonates. The 13C-NMR spectroscopy revealed the formation of random sequences in the poly(ester carbonate)s due to the carbonate interchange reactions. The number average molar mass of the polymers was in the range 4500 to 14700 g mol−1 with respect to the different diols used. The reaction of HBHOE with DEC yielded polyricinoleate due to a polytransesterification reaction. The fatty acid derivative-based polymers were amorphous in nature and displayed a glass transition well below the room temperature. The temperature at 10% weight loss, determined using thermogravimetric analysis in nitrogen atmosphere, of the polycarbonates and poly(ester carbonate)s was in the range 273 to 325 °C. A large difference between the glass transition (−58 to −75 °C) and initial decomposition temperature (176 to 226 °C) was observed. This offers the derived polymers a wide processing window.
Acknowledgements
The authors would like to thank University of Bordeaux-1, ANR, IPB, CNRS and Aquitaine Council for financial support of this research.References
- Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008 Search PubMed.
- Z. S. Petrović, Polym. Rev., 2008, 48, 109–155 CrossRef.
- H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2010, 112, 10–30 CrossRef CAS.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- H. Hojabri, X. Kong and S. S. Narine, Biomacromolecules, 2010, 11, 911–918 CrossRef.
- Y. S. Lu and R. C. Larock, Biomacromolecules, 2007, 8, 3108–3114 CrossRef CAS.
- Z. S. Petrovic, I. Cvetkovic, D. P. Hong, X. Wan, W. Zhang, T. Abraham and J. Malsam, J. Appl. Polym. Sci., 2008, 108, 1184–1190 CrossRef CAS.
- D. Quinzler and S. Mecking, Angew. Chem., Int. Ed., 2010, 49, 4306–4308 CrossRef CAS.
- U. Edlund and A. C. Albertsson, Adv. Drug Delivery Rev., 2003, 55, 585–609 CrossRef CAS.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- J. Tang, Z. Zhang, Z. Song, L. Chen, X. Hou and K. Yao, Eur. Polym. J., 2006, 42, 3360–3366 CrossRef CAS.
- E. Hablot, B. Donnio, M. Bouquey and L. Avérous, Polymer, 2010, 51, 5895–5902 CrossRef CAS.
- S. Cavus and M. A. Gurkaynak, Polym. Adv. Technol., 2006, 17, 30–36 CrossRef CAS.
- E. Zini, M. Scandola, Z. Jiang and R. A. Gross, Macromolecules, 2008, 41, 4681–4687 CrossRef CAS.
- Y. Tokiwa, Biopolymers, 2003, 9, 417–422 CAS.
- F. Hostettler and E. F. Cox, German Offen 116 1 545, Union Carbide Corp., 1970; F. Hostettler and E. F. Cox, German Offen 117 1 545, Union Carbide Corp., 1969; F. Hostettler and E. F. Cox, German Offen 118 1 545, Union Carbide Corp., 1969.
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1997, 30, 3470–3476 CrossRef CAS.
- Y. Hu, L. Qiao, Y. Qin, X. Zhao, X. Chen, X. Wang and F. Wang, Macromolecules, 2009, 42, 9251–9254 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- P. Pawłowski and G. Rokicki, Polymer, 2004, 45, 3125–3137 CrossRef.
- G. Rokicki, W. Kuran and J. Kielkiewicz, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 967–976 CrossRef CAS.
- P. Brignou, M. P. Gil, O. Casagrande, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS.
- R. A. Gross, B. Kalra and A. Kumar, Appl. Microbiol. Biotechnol., 2001, 55, 655–660 CrossRef CAS.
- V. Pokharkar and S. Sivaram, Polymer, 1995, 36, 4851–4854 CAS.
- Carbon Dioxide as a chemical Feedstock, ed. M. Aresta, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- S. D. Allen, C. M. Byrne and G. W. Coates, in Feedstocks for the Future, ACS Symp. Ser., 2006, 921, 116–129 Search PubMed.
- Z. S. Petrović, Polymers from Biological Oils Contemporary Materials, 2010, I–1, 39–50 Search PubMed.
- S. Chatti, G. Schwarz and H. R. Kricheldorf, Macromolecules, 2006, 39, 9064–9070 CrossRef CAS.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Biomacromolecules, 2010, 11, 1202–1211 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- B. G. Zanetti-Ramos, E. Lemos-Senna, V. Soldi, R. Borsali, E. Cloutet and H. Cramail, Polymer, 2006, 47, 8080–8087 CrossRef CAS.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- H. Adkins and R. H. Gillespie, Org. Synth., 1955, 3, 671 Search PubMed.
- R. S. Klonowski, T. W. Findley, C. M. Josephson and A. J. Striton, J. Am. Oil Chem. Soc., 1970, 47, 326–328 CrossRef CAS.
- B. A. Sweileh, Y. M. Al-Hiari, M. H. Kailani and H. A. Mohammad, Molecules, 2010, 15, 3661–3682 CrossRef CAS.
- Y. Jia, X. Shen, X. Gu, J. Dong, C. Mu and Y. Zhang, Polym. Adv. Technol., 2008, 19, 159–166 CrossRef CAS.
- M. Jayakannan and P. Anilkumar, J. Polym. Sci., Polym. Chem. Ed., 2004, 42, 3996–4008 CrossRef CAS.
- M. Yokoe, K. Aoi and M. Okada, J. Polym. Sci., Polym. Chem. Ed., 2003, 41, 2312–2321 CrossRef CAS.
- H. Ebata, K. Toshima and S. Matsumura, Macromol. Biosci., 2007, 7, 798–803 CrossRef CAS.
- I. C. McNeill and A. Rincon, Polym. Degrad. Stab., 1989, 24, 59–72 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra of oleyl carbonate, epoxidised oleyl carbonate and 4-hydroxybutyl-12-hydroxyoctadec-9-enoate (HBHOE). 13C-NMR spectra of ethyl(10(9)-hydroxy-9(10)-methoxyoctadecyl)carbonate (EHMOC), 4-[(ethoxycarbonyl)oxy]butyl-12-[(ethoxycarbonyl)oxy]octadec-9-enoate (EOBEOE), polycarbonate prepared by self-polycondensation of EHMOC (PC-I) and poly(ester carbonate) synthesized from EOBEOE with PEG200. 1H- and 13C-NMR spectra of poly(ester carbonate)s derived from EOBEOE with namely, hexanediol (PEC-III), PPG400 (PEC-V), PCL1250 (PEC-VI), isosorbide (PEC-VII) and polyricinoleate (PEC-VII) prepared from HBHOE and diethyl carbonate. MALDI-TOF spectra of PC-I, PEC-III, PEC-IV, PEC-V, PEC-VI, and PEC-VII. The DSC and TGA thermograms of PC and PECs. The SEC traces for polycarbonates and poly(ester carbonate)s. See DOI: 10.1039/c1py00326g |
| This journal is © The Royal Society of Chemistry 2011 |