Interfacial micellar phase transfer using amphiphilic invertible polymers†
Ivan
Hevus
,
Ananiy
Kohut
and
Andriy
Voronov
*
Department of Coatings and Polymeric Materials, North Dakota State University, Dept. 2760, P.O. Box 6050, Fargo, ND 58108-6050, USA. E-mail: andriy.voronov@ndsu.edu; Fax: +1 701 2318439; Tel: +1 701 2319563
First published on 13th October 2011
Abstract
Micelles formed from new amphiphilic invertible polymers (AIPs) can sequester hydrophobic molecules in water and transfer their payload to a non-polar phase. The amount of material transferred depends primarily on micellar loading capacity. Increasing the lipophilicity of the AIP composition increases the amount of material transferred. The obtained data indicate that AIPs have the potential to serve as an alternative for administration of polymer-based nanopharmaceuticals. Invertible polymeric micelles can potentially facilitate controlled release of poorly water-soluble agents incorporated within the micellar interior when compared to release by thermodynamically stable conventional (non-invertible) micelles.
Nanoscale therapeutic systems are considered as emerging novel modalities for cancer treatment.1–3 These systems include polymeric micelles that have several potential advantages such as high payload capacity, prolonged blood circulation times, reduced toxicity to healthy tissues and improved anti-tumor efficacy.4Polymer micellization and self-assembly are due to the mutual incompatibility of macromolecular fragments and specific interactions of polymer hydrophilic and lipophilic moieties with the solvent.5,6 The self-assembled polymeric micelles may act as containers and carriers that have potential in controlled and targeted delivery of drugs that are poorly soluble in water.4,7,8 By encapsulation of the drug within the hydrophobic core of the micelle, the apparent solubility of the drug can be significantly increased.8 Although, the utility of polymeric micelles in pharmaceutical formulations is now well recognized, the thermodynamic stability of conventional (block-copolymer) micelles in biological media often complicates the release of the active agents.9
In fact, upon entering the tumor site, it is desirable that the therapeutic agent is released from the micelles in a controlled way. Possible way to achieve this is to explore stimulus responsive drug release systems that include pH,10 temperature11 and ultrasound-stimulated release.12 Although polymeric micelles based on amphiphilic block copolymers are generally more stable and safer than conventional surfactant micelles,13 and have been studied as a drug carrier of a hydrophobic drug by hydrophobic interaction with the drug,13–18 to our knowledge, no micellar systems from polymers inducing release of the encapsulated in the micellar interior hydrophobic drug by rapid change of the environmental polarity are reported. Such release strategy is targeted in our research, when the responsive polymeric carrier with a hydrophobic payload can be transferred from aqueous environment to polar/non-polar interface and immediately release the payload upon inverting the macromolecular conformation by entering less polar environment.
We reported recently on a new class of polymers, amphiphilic invertible polymers (AIPs), that form micelles and self-assemble into micellar assemblies in response to polymer structure and concentration.19 The unique feature of AIP macromolecules is their ability for inverse conformational changes in response to changing solvent polarity. The invertibility of new polymers is especially promising for controlled self-assembly in applications that require utility simultaneously in polar and non-polar media, that includes drug delivery. It has been assumed that polymer fragments incompatibility achieved on a smaller length scale (in comparison to block copolymers) can allow greater tunability of the assemblies formation. The latter has been approached by synthesizing amphiphilic macromolecules containing precisely controlled number of hydrophilic and hydrophobic short fragments with a well-defined length. The control over the self-assembly additionally benefits from having the hydrophilic and hydrophobic fragments distributed alternately in the amphiphilic macromolecule.13 The amphiphilic invertible polymers differ from each other in hydrophilic–lipophilic balance (HLB), which considerably affects their surface activity and ability to self-assemble in polar and non-polar solvents. The AIP micellar assemblies behave like a host for otherwise insoluble molecules in polar (including aqueous) and non-polar solutions, and bind lipophilic and hydrophilic guest molecules, in water and toluene, respectively.19,20
The ability of amphiphilic invertible polymers to sequester lipophilic insoluble molecules in water and to switch conformation in response to polarity of the environment can be exploited to study the interactions between AIP micellar assemblies and biomembrane towards possible applications as nanopharmaceuticals. Adsorption of drug loaded micellar assemblies onto the cell membrane which is essentially a lipid matrix consisting of a phospholipid bilayer with inwardly oriented hydrophobic hydrocarbons, outwardly oriented hydrophilic heads,21 would be expected to change the AIP conformation. This could result in an enhancement of interactions between the polymer and membrane and subsequent release of the drug molecules. Probing this possibility requires that micellar assemblies first be confirmed to sequester poorly soluble guest molecules in aqueous solution and to transfer these molecules through polar/non-polar interface. Both aspects have been addressed in this work.
The chemical structure of the polymers 1–6 based on polytetrahydrofuran (PTHF) as a hydrophobic fragment, and poly(ethylene glycol) (PEG) as a hydrophilic fragment (Fig. 1 and Table 1) was confirmed by FTIR- and 1H NMR spectroscopic measurements (Fig. S1 in the ESI†). We targeted a broad surface activity for new polymers to achieve varying micellar capacity for sequestration of poorly soluble materials in water. To this end, the HLB of the synthesized macromolecules has been varied between 5 and 15 as calculated.22
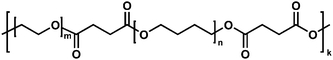 | ||
| Fig. 1 AIPs chemical structure. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000
000Nile red (7-diethylamino-3,4-benzophenoxazine-2-one) is a lipophilic dye that shows a solvatochromic behavior at a wide wavelength range.23Nile red is insoluble in water and shows no absorption in optical spectroscopy measurements (Fig. 2A, inset). However, in the presence of different amphiphilic polymers, namely 1, 3, and 5 (Table 1) (1% w/v), the dye was solubilized by AIP macromolecules dissolved in water, as shown in Fig. 2A and B. They provided a microenvironment that was capable of sequestering hydrophobic dye molecules. When Nile red was added to an aqueous solution of polymer 1, significantly lower absorption intensity in comparison to polymers 3 and 5 was observed (Fig. 2A and B). Although macromolecules in composition 1 did sequester the dye, a smaller number of hydrophobic molecules were solubilized. The latter effect can be explained by the higher hydrophobicity of polymers 3 and 5 in comparison to 1. The differences in HLB values also explain the marked difference in absorption intensity observed in dye sequestration by polymer 3 compared to 5.
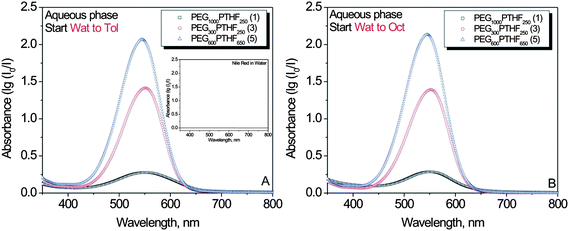 | ||
| Fig. 2 Optical spectra of Nile red-loaded polymers (1, 3, and 5) dissolved in water (1% w/v) (two experimental sets A and B). | ||
It was significantly higher when dye was solubilized by macromolecules with a lower HLB value. The data on sequestration of Nile red by AIPs were linked with measurements of the AIP critical micelle concentration (cmc). The cmc values of polymers 1, 3, and 5 were determined in a pyrene-solubilization study (Fig. S2 in the ESI†). Pyrene is a commonly used spectroscopic probe for studying polymeric micellization and self-assembly.24,25 In the present case, a significantly higher cmc value was observed for polymer 1 compared to 3 and 5 (Table 1). This result establishes the higher surface activity and stronger micellization of AIPs with lower HLB values and explains their better capacity for solubilizing hydrophobic molecules in aqueous solutions.
To probe the transfer of dye-loaded polymeric micelles through a polar/non-polar interface to a solvent in which Nile red is soluble, we chose two non-polar solvents (toluene and 1-octanol) that are immiscible with water. The non-polar solvent was added to the top of an aqueous solution containing dye-loaded polymeric micelles. The goal was to study the behavior of these micelles in the presence of two solvents with opposite polarity, and to answer the question whether the hydrophobic payload could be delivered from the aqueous phase to the non-polar organic phase. Being more polar than toluene, 1-octanol was used in our experiments as the most valid hydrophobicity scale model system to study partitioning in biomembrane.26 Experimental mixtures were shaken for one hour and then allowed to separate. The polymer-sequestered Nile red, normally insoluble in water, was successfully delivered to both toluene and 1-octanol using polymers 1, 3, and 5. All three AIPs were capable of transferring dye molecules from water to an organic medium of opposite polarity that is seen by top (organic) phase color appearance.
Due to the solvatochromic nature of Nile red, we noted the differences in transferred dye solution color in toluene (pink) and 1-octanol (orange) (Fig. 3A and B). In addition, a significant difference in dye solution color intensity was noted, both in 1-octanol and toluene, when different polymers were used for the transfer (Fig. 3C and D). We assume that the differences in dye solution color intensity can be attributed either to: (i) different loading capacity of polymeric micelles (ability to bind dye molecules in aqueous medium) and/or (ii) different transfer efficiency of the polymeric micelles (ability to deliver and release dye molecules from aqueous to organic non-polar phase). We distinguished between these two factors by comparing an initial concentration of the polymer-solubilized dye in water with the final Nile red concentration in toluene and 1-octanol after the transfer, measured by optical spectroscopy. Analysis of the dye concentration in aqueous (before the transfer) and organic (after the transfer) phases revealed that vast majority of the dye molecules was transferred to the toluene and 1-octanol. This observation obviously rules out the factor of different transfer efficiency of polymeric micelles. The ability of the polymer to bind dye molecules in water (loading capacity) was the major determinant of the extent of transfer of Nile red by polymeric micelles through the polar/non-polar interface.
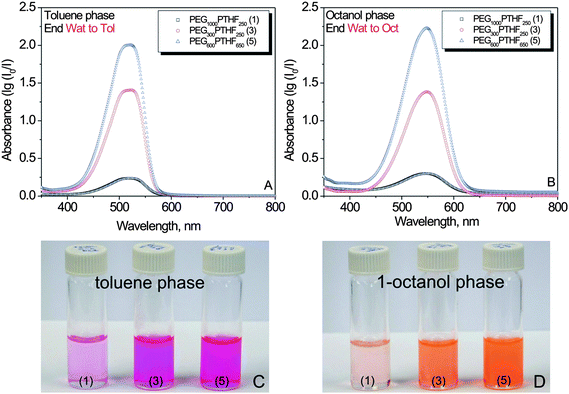 | ||
| Fig. 3 Optical spectra of Nile red solutions transferred by polymers 1, 3, and 5 to toluene (A) and 1-octanol (B). Appearance of dye solutions in toluene (C) and 1-octanol (D) after the polymer (1, 3, or 5) mediated-transfer. | ||
With the AIP-mediated dye transfer established, a mechanism for hydrophobic molecule release from polymeric micelles was considered as next. At least three different possibilities exist to explain how dye molecules could cross a polar/non-polar interface. One of these assumes that the molecules that are poorly soluble in water can be partitioned between the micellar interior and bulk water, due to their limited aqueous solubility.26 At the beginning of the experiment, Nile red molecules are distributed between the bulk water and the AIP micelles that act as containers of poorly soluble material. The limited solubility of Nile red in water, and thus, the low concentration of the dye in the aqueous phase, is sufficient to transfer the Nile red from the micellar interior, first to water and then from water to the non-polar phase (Fig. 4A).27
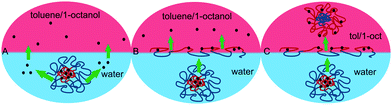 | ||
| Fig. 4 Possible mechanism of Nile red transfer and release from an aqueous to an organic (non-polar) phase. | ||
At the same time, the dye-loaded polymeric containers can migrate from the aqueous phase to the polar/non-polar interface, change their conformation (due to the polymer invertibility characteristic) upon changing polarity, disassemble (lose their containers properties) and release dye molecules into the non-polar solvent, where Nile red is soluble (Fig. 4B). Ultimately, the dye-loaded polymeric micelles can undergo thermodynamically driven distribution between the aqueous and organic phases. This can happen when dye-loaded micelles (due to the invertibility of macromolecule) cross the interface, invert their conformation, and form new inverted micelles in the non-polar phase (Fig. 4C). If this mechanism works, then dye-loaded AIP micelles act as carriers for solubilized Nile red molecules and may release the dye upon inversion within a non-polar phase.
To distinguish between the three possible mechanisms, we conducted an experiment to determine the presence of AIP macromolecules in toluene and 1-octanol phases. They were detected in both the toluene and 1-octanol systems for each polymer tested in the transfer experiment (Table 2) indicating that the AIP macromolecules are able to cross the interface between water and 1-octanol/toluene.
| AIP | HLB | cmc (wt%) | Fraction of transferred polymer | |
|---|---|---|---|---|
| To tol. | To 1-oct. | |||
| 1 PEG1000PTHF250 | 14.9 | 0.067 | 0.11 | 0.06 |
| 3 PEG300PTHF250 | 9.9 | 9.4 × 10−4 | 0.29 | 0.26 |
| 5 PEG600PTHF650 | 9.3 | 3.5 × 10−4 | 0.10 | 0.14 |
In summary, we have shown that: (i) AIP polymeric micelles can sequester hydrophobic molecules in aqueous solutions, cross polar/non-polar liquid interface and transfer their payload to the non-polar phase of an immiscible solvent mixture. The micelles are stable in water and exhibit both container and carrier properties while transferring the material; (ii) the amount of transferred material depends mainly on micellar loading capacity (the ability of the AIP macromolecules making up the micelles to bind guest molecules). Increasing the lipophilicity of the AIP increases the amount of transferred material; and (iii) the mechanism of material release involves the AIP micelles crossing the polar/non-polar interface and the partitioning of the poorly soluble material between the micellar interior and bulk water, due to material limited solubility.
The findings reported here indicate that AIPs have the potential to serve as alternatives for the nanopharmaceuticals based on existing polymeric micelles. It is known that the thermodynamic stability of conventional block-copolymer (non-invertible) micelles in biological media often complicates the release of the active agents when the micellar carriers enter the cell interior. We showed that polymeric micelles from AIPs can overcome this complication by undergoing invertible changes in macromolecular conformation in response to changes in polarity. This may facilitate controlled release of poorly soluble agents incorporated within the micellar interior. Further explorations of the transfer mechanism and applications of AIPs as new alternatives for nanopharmaceutical delivery are presently underway in our laboratory.
Acknowledgements
Partial support for this work from the North Dakota EPSCoR/National Science Foundation (EPS-0447679) and National Science Foundation (CBET-0966574) is gratefully acknowledged.References
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- S. R. Croy and G. S. Kwon, Curr. Pharm. Des., 2006, 12, 4669–4684 CrossRef CAS; N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef; D. Sutton, N. Nasongkla, E. Blanco and J. Gao, Pharm. Res., 2007, 24, 1029–1046 CrossRef; V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 Search PubMed; M. Yokoyama, Expert Opin. Drug Delivery, 2010, 7(2), 145–158 CrossRef; G. Kwon, Crit. Rev. Ther. Drug Carrier Syst., 2003, 20(5), 357–403 CrossRef.
- D. F. Evans and H. Wennerstrom, The Colloidal Domain, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- T. S. Kale, A. Klaikherd, B. Popere and S. Thayumanavan, Langmuir, 2009, 25(17), 9660–9670 CrossRef CAS.
- S. Ghosh, K. Irvin and S. Thayumanavan, Langmuir, 2007, 23, 7916–7919 CrossRef CAS.
- H. Montazeri and A. Lavasanifar, Expert Opin. Drug Delivery, 2006, 3(1), 139–162 CrossRef.
- W. E. Bawarski, E. Chidlowsky, D. J. Bharali and S. A. Mousa, Nanomed.: Nanotechnol., Biol. Med., 2008, 4, 273–282 CrossRef CAS.
- Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16, 122–130 CrossRef CAS; A. Potineni, D. M. Lynn, R. Langer and M. M. Amiji, J. Controlled Release, 2003, 86, 223–234 CrossRef; H. R. Stapert, N. Nishiyama, D. L. Jiang, T. Aida and K. Kataoka, Langmuir, 2000, 16, 8182–8188 CrossRef; Y. Tang, S. Y. Liu, S. P. Armes and N. C. Billingham, Biomacromolecules, 2003, 4, 1636–1645 CrossRef.
- S. Q. Liu, Y. W. Tong and Y. Y. Yang, Mol. BioSyst., 2005, 1, 158–165 RSC; J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS.
- S. Mitragotri, Nat. Rev. Drug Discovery, 2005, 4, 255–260 CrossRef CAS; Z. G. Gao, H. D. Fain and N. Rapoport, J. Controlled Release, 2005, 102, 203–222 CrossRef; J. D. Priutt and W. G. Pitt, Drug Delivery, 2002, 9, 253–258 CrossRef.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109(1–3), 169–188 CrossRef CAS.
- I. Astafieva, X. F. Zhong and A. Eisenberg, Macromolecules, 1999, 26(26), 7339–7352 CrossRef.
- V. P. Torchilin, J. Controlled Release, 2001, 73(2–3), 137–172 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- D. Le Garrec, M. Ranger and J. C. Leroux, Am. J. Drug Delivery, 2004, 2, 15–42 CrossRef CAS.
- C. F. Van Nostrum, Adv. Drug Delivery Rev., 2004, 56, 9–16 CrossRef CAS.
- A. Kohut, A. Voronov, O. Gevus and W. Peukert, Langmuir, 2006, 22, 1946 CrossRef CAS; A. Voronov, A. Kohut and W. Peukert, Langmuir, 2007, 23, 360 CrossRef; A. Voronov, A. Kohut and S. Vasylyev, et al. , Langmuir, 2008, 24, 12587 CrossRef; L. Martinez Tomalino, A. Voronov, A. Kohut and W. Peukert, J. Phys. Chem. B, 2008, 112, 6338 CrossRef; A. Kohut and A. Voronov, Langmuir, 2009, 25, 4356 CrossRef.
- A. Kohut, L. Sieburg, S. Vasylyev, O. Kudina, I. Hevus, S. Stafslien, J. Daniels, V. Kislenko and A. Voronov, Amphiphilic Invertible Polymers (AIPs). Micellization and Self-Assembly in Aqueous Solutions, in Amphiphiles: Molecular Assembly and Applications, ACS Symposia Series Book, Washington DC, 2011, pp. 209–224 Search PubMed.
- S. J. Singer and G. L. Nicolson, The fluid mosaic model of the structure of cell membranes, Science, 1972, 175, 720 CAS.
- J. T. Davies and E. K. Rideal, Interfacial Phenomena, Academic Press, New York, 1961, p. 371 Search PubMed.
- J. F. Deye and T. A. Berger, Anal. Chem., 1990, 62, 615–622 CrossRef CAS.
- L. Sieburg, A. Kohut, V. Kislenko and A. Voronov, J. Colloid Interface Sci., 2010, 351(1), 116–121 CrossRef CAS.
- C. Schmitz, A. Mourran, H. Keul and M. Möller, Macromol. Chem. Phys., 2008, 209, 1859–1871 CrossRef CAS.
- A. Berthod and C. Garcia-Alvarez-Coque, Micellar Liquid Chromatography, CRC Press, 1st edn, 2000 Search PubMed.
- S. Basu, D. R. Vutukuri and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 16794–16795 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Text and figures giving experimental details regarding the amphiphilic invertible polymers synthesis and characterization. See DOI: 10.1039/c1py00399b |
| This journal is © The Royal Society of Chemistry 2011 |