Synthesis of end-functionalized poly(N-isopropylacrylamide) with group of asymmetrical phthalocyaninevia atom transfer radical polymerization and its photocatalytic oxidation of Rhodamine B
Zhengguo
Gao
a,
Xiangdong
Tao
a,
Yuan
Cui
a,
Toshifumi
Satoh
b,
Toyoji
Kakuchi
b and
Qian
Duan
*a
aSchool of Materials Science and Engineering, Changchun University of Science and Technology, Changchun, 130022, China. E-mail: duanqian88@hotmail.com; Fax: +86 431 85306769; Tel: +86 431 85583105
bDivision of Biotechnology and Macromolecular Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan
First published on 26th September 2011
Abstract
A novel asymmetrical zinc(II) phthalocyanine (ZnPcBr) was synthesized as the initiator for atom transfer radical polymerization (ATRP). Using CuBr/tris(2-dimethylaminoethyl)amine (Me6TREN) as the catalyst system, the ATRP of N-isopropylacrylamide (NIPAM) was carried out to afford a new linear poly(N-isopropylacrylamide) with end group of asymmetrical phthalocyanine (ZnPc-PAM). The structures of the initiator and the polymers were characterized by means of FT-IR and 1H NMR. Polydispersity index (PDI) obtained by gel permeation chromatography (GPC) indicated that the molecular weight distribution was narrow. The lower critical solution temperatures (LCST) for the aqueous solutions of ZnPc-PAM, measured by turbidimetry method and differential scanning calorimetry (DSC), respectively, were increased due to the incorporation of the phthalocyanine group at the chain end and lowered with increasing molecular weight. It was verified that ZnPc-PAM possessed photocatalytic activity by the degradation of Rhodamine B in the presence of hydrogen peroxide under visible light. Moreover, it presented the highest catalytic efficiency at around its LCST owing to the thermo-sensitive property, which benefited the recycling of the polymeric photocatalyst.
1. Introduction
Phthalocyanines are planar aromatic macrocycles consisting of four isoindole units presenting an 18 π-electron aromatic cloud delocalized over an arrangement of alternated carbon and nitrogen atoms. They have been investigated extensively and used in many fields, including chemical sensors, photodynamic therapy, electrochromic displays, catalysts for oxidative degradation, information storage systems, nonlinear optics, etc.1–7 Especially in the field of photocatalytic oxidation, the metallophthalocyanines have attracted considerable attention due to their high electron transfer abilities and redox properties as well as the thermal and chemical stability. They have been used for catalytic oxidation of alkanes, olefins, aromatic and other compounds through oxidase-like, catalase-like and peroxidase-like mechanisms.8–12 However, their applications were restricted by the following problems: poor solubility, high tendency for aggregation and difficulty in recycling. The solubility in solution, especially in water, could be improved by incorporating suitable substitution on the peripheral circle of phthalocyanine. Immobilizing the phthalocyanine complexes onto insoluble solid substrates could avoid the formation of aggregation and facilitate the recycling of the catalyst. However, there were several shortcomings in these strategies, for instance, the substituting groups were limited, and the transmittance was decreased due to the using of the insoluble supports.Poly(N-isopropylacrylamide) (PNIPAM) is the most extensively studied thermo-responsive polymer, which is clear at room temperature and able to give a reversible phase transition at ca. 32 °C (lower critical solution temperature, LCST).13–15 It has been widely used in many fields, such as drug delivery systems, separation and purification, nanotechnology and bioengineering.16–21 The thermo-responsive property could be used to facilitate recycling of the catalyst by simple heating and filtration. By combining PNIPAM with phthalocyanine derivatives, the polymer possessed both the catalytic activity and the thermo-responsive property, which would avoid the aggregation of the phthalocyanine in solution, and facilitate the recycling of the catalyst. Chen and co-workers reported the preparation of several novel thermo-sensitive photocatalysts by grafting (co)polymerizing with N-isopropylacrylamide (NIPAM) from the metallophthalocyanine derivatives.22 The investigations showed the polymeric photocatalysts possessed high catalytic activity for the oxidation of phenols and thiols under visible light irradiation. However, the structure of the polymer prepared by traditional radical polymerization is not well-defined, and its molecular weight distribution is wide, which will affect the property accordingly. Recently, controlled/“living” radical polymerization (CRP) has been widely developed, which provides an efficient way of synthesizing polymers with well-defined structure and narrow molecular weight distribution. Atom transfer radical polymerization (ATRP), one of the most investigated CRP methods, can provide polymers with designed structure using suitable initiators.23–26 For instance, Kimura et al. reported the formation of amphiphilic phthalocyanines with substitution of polyacrylates through ATRP, which self-assembled into fibrous aggregates in methanol.27 Recently, our group synthesized several end-functionalized PNIPAMs and the effect of end group on the thermo-responsive property was also studied.28–30
However, few studies of ATRP of NIPAM initiated from the phthalocyanine derivatives were reported. Herein, we presented the synthesis of the end-functionalized PNIPAM with asymmetrical zinc(II) phthalocyaninevia the ATRP technique using ZnPcBr as the initiator and CuBr/Me6TREN as the catalyst system at 70 °C. The polymer with narrow molecular weight distribution possessed both thermo-responsive property and photocatalytic activity. The changes of LCSTs of ZnPc-PAM caused by the incorporation of phthalocyanine end group were studied. We then tested the photocatalytic activity on the oxidation of Rhodamine B (Rh B) under the visible light irradiation.
2. Experimental
2.1 Materials
N-Isopropylacrylamide (99%, Aldrich) was recrystallized twice from benzene/hexane (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) prior to use; 2-bromoisobutyryl bromide (BiBB, 99%, Acros); copper(I) bromide (CuBr, 99%, Aldrich); 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Alfa Aesar); cellophane tube (MWCO 2000, Solarbio); tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized according to the literature;31 the precursors 4,5-bis(decyloxy)phthalonitri, 4-methoxy-5-((2-(tetrahydropyran-2-yl)oxy)ethoxy)phthalonitrile were synthesized by the literature methods respectively.32,33 All other chemicals were purchased from Sinopharm Chemical Reagent Co. and were used as received.
:1, v/v) prior to use; 2-bromoisobutyryl bromide (BiBB, 99%, Acros); copper(I) bromide (CuBr, 99%, Aldrich); 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Alfa Aesar); cellophane tube (MWCO 2000, Solarbio); tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized according to the literature;31 the precursors 4,5-bis(decyloxy)phthalonitri, 4-methoxy-5-((2-(tetrahydropyran-2-yl)oxy)ethoxy)phthalonitrile were synthesized by the literature methods respectively.32,33 All other chemicals were purchased from Sinopharm Chemical Reagent Co. and were used as received.
2.2 Characterization
All nuclear magnetic resonance (NMR) spectra were recorded on a Unity 400 (Varian) NMR spectrometer with CDCl3 as the solvent. Fourier transform infrared (FT-IR) spectra were recorded on a FTIR 8400S (Shimadzu) spectrometer. The spectra were collected using 40 scans with a resolution of 4 cm−1. Molecular weights and molecular weight distributions were determined by gel permeation chromatography (GPC) equipped with Waters 600 pump and Waters 410 differential refractometer at 25 °C. The eluent was THF at a flow rate of 1.0 mL min−1. A series of low polydispersity polystyrene standards were employed for GPC calibration. Ultraviolet-visible (UV-vis) spectra were measured on a UV mini 1240 (Shimadzu) spectrophotometer. Differential scanning calorimetry (DSC) measurements were carried out on a Perkin-Elmer DSC Instrument at a heating rate of 10 °C min−1 in flowing nitrogen.2.3 Synthesis of zinc(II) 2-hydroxyethoxy-3-methoxy-9,10,16,17,23,24-hexakis(decyloxy)phthalocyanine (ZnPcOH)
A mixture of 4-methoxy-5-((2-(tetrahydropyran-2-yl)oxy)ethoxy)phthalonitrile (1.1 g, 3.4 mmol), 4,5-bis(decyloxy)phthalonitrile (4.4 g, 10 mmol), ZnCl2 (0.82 g, 6 mmol) and DBU (1 mL) in n-pentanol (30 mL) was stirred and slowly heated. The mixture was refluxed under an argon atmosphere for 50 h. After the mixture was cooled, methanol (150 mL) was added, and the resulting precipitate was filtered off. The precipitate was treated with p-toluenesulfonic acid monohydrate (1.5 g) in a mixed solution of methanol and CH2Cl2 (50 mL, 1:2 v/v) to remove tetrahydro-2H-pyran. After removal of the solvent, the residue was washed off with methanol. The crude product was purified by column chromatography (SiO2, CHCl3/methanol 9:1 v/v). 82 mg, yield: 15%. FT-IR (KBr): 3425 cm−1 (υOH), 1604, 1496 cm−1 (υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C); 1H NMR (400 MHz, CDCl3): δ = 8.19 (br s, 8H, ArH), 5.74 (br s, 1H, OH), 4.58 (m, 12H, ArOCH2), 3.98 (t, 2H, CH2OH), 3.45 (s, 3H, ArOCH3), 2.65(s, 2H, ArOCH2), 2.18 (br t, 12H, OCH2CH2), 1.2–1.7 (m, 84H, CH2), 0.90 (t, 18H, CH3).
C–C); 1H NMR (400 MHz, CDCl3): δ = 8.19 (br s, 8H, ArH), 5.74 (br s, 1H, OH), 4.58 (m, 12H, ArOCH2), 3.98 (t, 2H, CH2OH), 3.45 (s, 3H, ArOCH3), 2.65(s, 2H, ArOCH2), 2.18 (br t, 12H, OCH2CH2), 1.2–1.7 (m, 84H, CH2), 0.90 (t, 18H, CH3).
2.4 Synthesis of zinc(II) 2-(2-bromoisobutyryloxyester)ethoxy-3-methoxy-9,10,16,17,23,24-hexakis(decyloxy)phthalocyanine (ZnPcBr)
The initiator ZnPcBr was synthesized using a reported method27 as shown in Scheme 1. To a solution of ZnPcOH (50 mg, 31 μmol) and triethylamine (1.5 mL) in CH2Cl2 (30 mL), BiBB (0.35 g, 1.5 mmol) was added in an ice bath under a nitrogen atmosphere. The mixture was stirred for 2 h at 0 °C and then one night at room temperature. The formed precipitate triethylamine hydrobromide was filtered off. After the solution was removed, the crude product was purified by column chromatography (SiO2, CHCl3). 44 mg, yield: 81%. FT-IR (KBr): 1739 cm−1 (υCO); 1H NMR (400 MHz, CDCl3): δ = 8.91 (s, 8H, ArH), 4.59 (m, 12H, ArOCH2), 4.17 (s, 2H, CH2OC(O)), 3.83 (s, 3H, ArOCH3), 2.77 (s, 2H, ArOCH2), 2.12 (br, 12H, OCH2CH2), 1.36–1.73 (m, 84H, CH2), 0.89 (t, 18H, CH3).
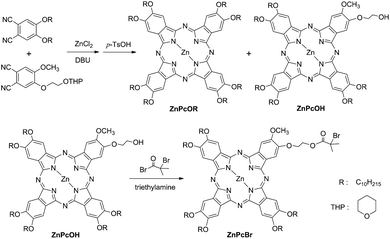 | ||
| Scheme 1 Synthetic route for the preparation of initiator ZnPcBr. | ||
2.5 General procedure for the end-functionalized PNIPAM with phthalocyanine group (ZnPc-PAM) viaATRP
The ATRP of NIPAM was performed at 70 °C in toluene. A general procedure employed for the preparation of ZnPc-PAM was as follows (shown in Scheme 2). A solution of NIPAM (1.7 g, 15 mmol), Me6TREN (27.5 μL, 0.1 mmol) and toluene (5 mL), and CuBr (14.5 mg, 0.1 mmol) was deoxygenated by bubbling with nitrogen and combined. After three freeze-pump-thaw cycles were performed, the initiator ZnPcBr (0.175 g, 0.1 mmol), dissolved in degassed toluene (1 mL), was added to begin the polymerization. The reaction was carried out at 70 °C for a certain time under a nitrogen atmosphere. Polymerization was terminated by exposure to air. Then the reaction mixture was diluted with THF and passed through an alumina column to remove the copper complex. The resulting polymer was purified by dialysis using a cellophane tube (MWCO, 2000) in DMF. After removing the solvents, the residue was dried in vacuo for 24 h to give the polymer ZnPc-PAM. 1H NMR (400 MHz, CDCl3): δ = 8.39 (ArH), 6.19 (NH), 3.99 (CH), 3.01 (ArOCH3), 2.11 (CH2CH), 1.88 (CH2), 1.62 (CH2), 1.43 (OCH2CH2), 1.14 (CH3), 0.85 (CH3).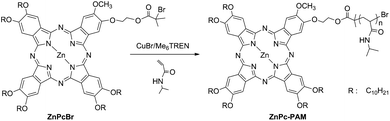 | ||
| Scheme 2 Synthetic route for the well-defined ZnPc-PAMviaATRP. | ||
2.6 Cloud points measurement
Cloud points (CP) of aqueous solutions of ZnPc-PAM were measured by UV-vis spectroscopy. Transmittance of the solution was recorded with a photodidode connected to a computer. The sample, whose concentration in distilled water was 2 mg mL−1, was poured into a 1 cm cell. The cell holder in the spectrophotometer was thermally controlled. The change in transmittance at 500 nm was monitored by heating the solution at a rate of 0.5 °C min−1.2.7 Photocatalytic degradation test
Catalytic oxidation of Rh B was carried out in aqueous solution at room temperature using hydrogen peroxide (H2O2) as the oxidant under visible light irradiation (using a 500 W halogen lamp through a glass filter (λ > 450 nm) as the irradiation source). The pH value of the solution was adjusted by adding standard buffer solutions, and the initial concentration of Rh B solution was 5 × 10−5 mol L−1. After the polymeric catalyst ZnPc-PAM (Mn, GPC = 5900, Mw/Mn = 1.18) was added into the Rh B solution under stirring, the samples (3 mL) were monitored immediately at given time intervals on a UV-vis spectrometer.3. Results and discussion
3.1 Synthesis and characterization
The initiator ZnPcBr was synthesized by the reaction of BiBB with the hydroxy group in the asymmetrical phthalocyannine ZnPcOH. It was worth mentioning that the asymmetrically substituted phthalocyanines were more difficult to obtain and the yields were low. Besides the hydroxy protection by the tetrahydorpyran (THP) group, we used a strong organic base DBU as the catalyst in order to increase the yield. After the separation of by-products by column chromatography, the monohydroxy substituted phthalocyanine ZnPcOH was obtained, and the yield was increased to 15% (with respect to dicyanide). The FT-IR spectra of ZnPcOH and ZnPcBr are shown in Fig. 1(a and b). After the esterification reaction, there was a strong absorption peak at 1737 cm−1 assigned to the carbonyl stretching vibration and the strong absorbance characteristic of hydroxyl group at 3431 cm−1 almost disappeared. The absorption peaks at 1601 and 1502 cm−1 were attributed to the skeletal vibration of the benzene rings in phthalocyanine. The 1H NMR spectrum of ZnPcBr and the corresponding peak assignments are shown in Fig. 2(a). All signals characteristic of both the phthalocyanine skeleton and the peripheral substations can be clearly observed. | ||
| Fig. 1 FT-IR spectra of (a) ZnPcOH, (b) ZnPcBr and (c) ZnPc-PAM. | ||
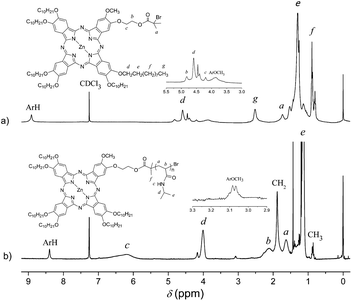 | ||
| Fig. 2 1H NMR spectra of (a) ZnPcBr and (b) ZnPc-PAM in CDCl3. | ||
To the best of our knowledge, there were no reports on the ATRP of NIPAM with asymmetrical phthalocyanine derivatives as the initiator. In the present work, ATRP of NIPAM was carried out in toluene at 70 °C using ZnPcBr as the initiator and CuBr/Me6TREN as the catalyst system. The feed ratio of [NIPAM]0:[ZnPcBr]0:[CuBr]0:[Me6TREN]0 was 150:1:1:1. Table 1 summarized the results of the polymerization reactions. Considering the effect of the end group, the molecular weights of the polymer were relatively low. The relative molecular weight and the polydispersity index (PDI) were obtained through GPC. The PDIs remained narrow with values between 1.14 and 1.20, which revealed the polymerization of NIPAM was in a controlled process.34,35
| Sample | Time (h) | Conv.b (%) | M n c | M w c | PDIc | LCST d (°C) |
|---|---|---|---|---|---|---|
|
a
[NIPAM]0:[ZnPcBr]0:[CuBr]0:[Me6TREN]0 = 150:1:1:1; solvent: toluene; temp.: 70 °C.
b Determined by gravimetric measurement.
c Determined by GPC using THF as eluent relative to polystyrene standards.
d Measured by turbidimetry using UV-vis spectrophotometer.
|
||||||
| P1 | 1.5 | 12 | 3890 | 4470 | 1.15 | 37.9 |
| P2 | 2.5 | 19 | 4970 | 5950 | 1.20 | 35.6 |
| P3 | 3.0 | 24 | 5900 | 6960 | 1.18 | 34.3 |
| P4 | 4.0 | 29 | 6720 | 7860 | 1.17 | 33.6 |
| P5 | 6.0 | 43 | 9010 | 10250 | 1.14 | 32.8 |
After a series of purifications, GPC traces of ZnPc-PAM (shown in Fig. 3) were relatively symmetric and showed no tailing at either side, suggesting the absence of any small molecular residues in the final product, such as the initiator, monomer or other byproducts. 1H NMR spectrum of ZnPc-PAM and the corresponding peak assignments are shown in Fig. 2(b). All signals characteristic of the repeating unit NIPAM can be clearly observed. Moreover, the signals appear at 9.49 and 1.88 ppm assigned to the end group of phthalocyanine. 1H NMR signals associated with the terminal 2-bromopropionate residues in ZnPcBr are clearly discernible at 0.85 ppm (methyl proton). The molecular weight could be estimated by comparing the resonance signals in the aromatic part of the terminal ZnPc group (δ = 8.39 ppm) with those of the methine protons of NIPAM (δ = 3.99 ppm). The molecular weight of ZnPc-PAM (Mn,GPC = 5900) determined by the 1H NMR analysis was 5820, which was in relatively good agreement with that determined by the GPC analysis.
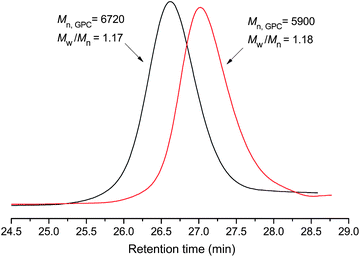 | ||
| Fig. 3 GPC traces of ZnPc-PAM obtained though ATRP. | ||
The structure of ZnPc-PAM was also characterized by FT-IR spectrum (as shown in Fig. 1(c)). The absorption peaks characteristic of PNIPAM can be clearly observed, as evidenced by the presence of a carbonyl stretching vibration (the secondary amide I band, υCO) with a weak red-shift to 1643 cm−1 and the N–H bending vibration (the secondary amide II bond band, δN–H) at 1548 cm−1. The strong absorbance at 3477 cm −1 was assigned to the stretching vibration (υN–H) of the acylamino group. The peaks of the skeletal vibration characteristic of the benzene rings in phthalocyanine at 1604 and 1502 cm−1 were overlapped by the amide bands. Thus, it was suggested that the well-defined ZnPc-PAM with end group of the asymmetrical phthalocyanine was successfully prepared though ATRP.
It is known that the phthalocyanine derivatives tend to aggregate in solution, which has direct influence on their properties. The aggregation behaviors of the phthalocyanine complexes can be revealed by the electronic absorption spectra.36,37Fig. 4(a) shows the UV-vis spectra of ZnPcBr and ZnPc-PAM in CH2Cl2 solution. It is clearly seen that both have the characteristic absorption bands (called B-band and Q-band) of the phthalocyanine derivatives in the UV-vis range. The maximum absorption wavelength (λmax) of the latter is located at around 709 nm, which was somewhat blue-shifted relative to that of the initiator ZnPcBr due to the combination with PNIPAM. Besides, the solubility of ZnPc-PAM was improved, which could dissolve in water and common organic solvents. As can be seen from the UV-vis spectra of ZnPc-PAM in aqueous solution with different concentrations, the Q-bands were not broadened and no shoulder peaks appeared with the increase of the concentrations (Fig. 4(b)). It could be concluded that aggregations of the phthalocyanine complex were avoided efficiently by being incorporated into PNIPAM.
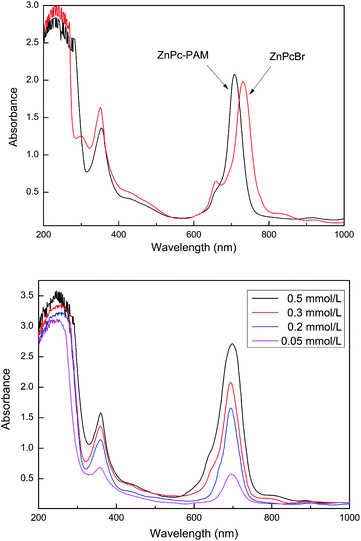 | ||
| Fig. 4 (a) UV-vis spectra of ZnPcBr and ZnPc-PAM in CH2Cl2 (3 × 10−4 mol L−1) and (b) UV-vis spectra of ZnPc-PAM in aqueous solution with different concentrations. | ||
3.2 Thermo-responsive property of ZnPc-PAM
Measuring CP is the simplest, most convenient method of determining the LCST of PNIPAM. CP of aqueous solutions of the phthalocyanine-terminated PNIPAM was measured by UV-vis spectroscopy. The changes in transmittance at 500 nm were recorded by heating the solution at a rate of 0.5 °C min−1. The aqueous solution of ZnPc-PAM was clear at low temperature and became turbid by raising the temperature. Fig. 5 shows the temperature dependence of CP for ZnPc-PAM in aqueous solution. It is obvious that there is a sharp transition in the CP curve, which indicates the thermo-responsive property of ZnPc-PAM. In the current study, the LCST was arbitrarily defined as the temperature corresponding to a 10% decrease of transmittance. The LCSTs of the aqueous solutions of P1–P5 with Mn,GPC of 3890, 4970, 5900, 6720 and 9010 were 37.9, 35.6, 34.3, 33.6 and 32.8 °C, respectively (as listed in Table 1), which were higher than that of NIPAM homopolymers. This was not consistent with the reports that the hydrophobic end groups decreased the thermal phase transition temperature of PNIPAM.35 It was attributed to the association of two aspects. Firstly, ZnPc-PAM could form a hydrophobic core of phthalocyanine and a diffuse corona of PNIPAM chains to facilitate the micellization, which would isolate effectively very hydrophobic groups from water and hence dramatically suppress their hydrophobic effects.39,40 Secondly, the steric hindrance effect also played an important role. The tendency of ZnPc-PAM towards packed globules arising from the temperature increase was hindered by the huge planar structure and the long alkoxy substitutions on the peripheral benzene rings of phthalocyanine. The combined effects resulted in the enhancement of the LCST on the whole. However, the LCST values lowered and approached around 32 °C, commonly reported for PNIPAM, with the increase of molecular weight. The inverse molecular weight dependence of the cloud points was in agreement with the reports on the effect of hydrophobic end groups on the thermo-responsive property of PNIPAM.38 This was ascribed to the end group effect on the performance of polymer, which was more remarkable relatively for the low molecular weight samples. When the molecular weight increased, the proportion of phthalocyanine end group in the polymer chain lowered and the end effect of phthalocyanine group on ZnPc-PAM became less significant. Consequently, the LCST of ZnPc-PAM decreased with the increasing of the molecular weight.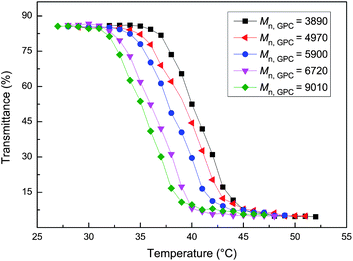 | ||
| Fig. 5 Temperature dependence of optical transmittance at wavelength of 500 nm obtained for aqueous solutions of ZnPc-PAM (2 mg mL−1). | ||
The thermal phase transitions of ZnPc-PAM were also measured by DSC. The DSC curve of ZnPc-PAM (Mn,GPC = 5900, Mw/Mn = 1.18) in aqueous solution was shown in Fig. 6. There was an obvious endothermic peak at 36.1 °C, which indicated a phase transition at this temperature. It is generally in good agreement with the LCST values determined by the turbidimetry method. The polymer chains absorbed energy, which caused the conformation change, and it appeared as precipitation from the solution in a macroscopic view.
 | ||
| Fig. 6 DSC traces for the aqueous solutions (10 mg mL−1) of ZnPc-PAM (Mn,GPC = 5900, Mw/Mn = 1.18). | ||
3.3 Measurement of photocatalytic degradation
Rhodamine B, a kind of commercial non-biodegradable toxic dye with aromatic structure, was employed to investigate the catalytic performance of ZnPc-PAM under the visible light. The degradation of Rh B in aqueous solution was estimated by the changes of the absorption intensity at the maximum absorption wavelength (around 550 nm). The degradation rate (D) can be defined as the ratio of the absorbance decrement to the initial of the Rh B aqueous solution as described as follows,where A0 is the initial absorption intensity at 550 nm, and A is the absorption intensity at any time during the measurement.

The photocatalytic activities of metallophthalocyanine were investigated in detail in the degradation of organic pollutants by H2O2 in previous works.41–43 To verify whether the polymers prepared possess the catalytic activity, controlled experiments of the degradation of Rh B were carried out. The results of the experiments at different conditions are displayed in Fig. 7. Rh B was hardly degraded in the presence of only ZnPc-PAM under visible light (curve d), and a similar phenomenon was observed in the presence of ZnPc-PAM and H2O2 without light irradiation (curve c). More than 81% of Rh B were degraded by H2O2 under visible light in the presence of ZnPc-PAM after 180 min (curve a), while limited oxidation occurred by H2O2 under the visible light (curve b). The degradation of Rh B was markedly accelerated by adding ZnPc-PAM to the system. The oxidation efficiency was improved obviously in the presence of ZnPc-PAM. Thus, we could conclude that the polymer ZnPc-PAM possessed high photocatalytic activity as expected.
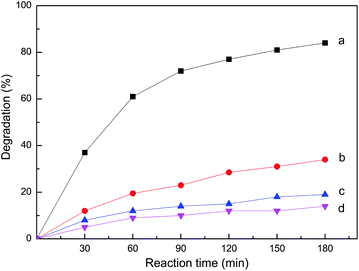 | ||
| Fig. 7 Comparison of different conditions for degradation of Rh B at 25 °C. a: ZnPc-PAM + H2O2 + visible light; b: H2O2 + visible light; c: ZnPc-PAM + H2O2; d: ZnPc-PAM + visible light. | ||
The concentration of Rh B declined gradually with increasing reaction time in the Rh B/ZnPc-PAM/H2O2 system, as confirmed by the UV-vis spectral changes of Rh B (Fig. 8). There were no other absorption peaks around 550 nm and the characteristic absorption band decreased in size rapidly and disappeared after degradation of 210 min. Concomitantly, the color of the reaction solution changed from the initial pink-red to nearly colorless transparent as shown in the inset of Fig. 8 (left). On the other hand, the approximately 709 nm bands were almost unchanged with increasing reaction time (inset of Fig. 8, right), suggesting the polymeric catalyst was relatively stable during the photodegradation of Rh B.
![UV-vis spectral changes of RhB in aqueous solution (initial concentration: 5 × 10−5 mol L−1) as a function of irradiation time. [ZnPc-PAM] = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4.](/image/article/2011/PY/c1py00308a/c1py00308a-f8.gif) | ||
| Fig. 8 UV-vis spectral changes of RhB in aqueous solution (initial concentration: 5 × 10−5 mol L−1) as a function of irradiation time. [ZnPc-PAM] = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4. | ||
Usually, temperature has less effect on the catalytic activity of the metallophthalocyanine derivatives. Considering the polymeric catalyst possessed thermo-responsive property, we investigated the effect of temperature on the oxidative degradation of Rh B. Fig. 9 showed the dependence of the catalytic activity of ZnPc-PAM on the reaction temperature within 50 min at pH 4. The Rh B aqueous solution was homogeneous and the degradation rate showed little change before 33 °C, which was evidently going up with the further increase of temperature. It reached peak value of 58% at 36 °C from 50% at 33 °C. Dramatically, the degradation rate declined when continuously raising temperature. The sudden change point was slightly lower than the LCST of ZnPc-PAM determined above. The interesting phenomenon was similar to the study on the photocatalytic degradation of 2-mercaptoethanol by Co-TACAPc/NIPAM copolymer reported by Chen.22,44 It was attributed to the thermo-responsive property of the polymeric catalyst. When the system temperature approached the LCST of ZnPc-PAM, the polymer chain turned to tightly packed globules from random coils to form a micelle-like structure. In the micelle-like area, the concentration of the active center zinc phthalocyanine was relatively higher, Rh B was oxidized efficiently. In addition, the degradation of Rh B was accelerated by heating continuously, for the oxidation reaction was an endothermic process.45 Due to the thermo-sensitivity, ZnPc-PAM precipitated from the aqueous solution when the temperature was raised successively. The system became heterogeneous and the active center zinc phthalocyanine was embedded by the PNIPAM chains, which hindered the axial coordination between the oxidant H2O2 and the center metal atom Zn in phthalocyanine. The catalytic activity declined and the degradation rate descended correspondingly.
![Effect of temperature on the degradation of Rh B in aqueous solution. [Rh B] = 5 × 10−5 mol L−1, [ZnPc-PAM] = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4.](/image/article/2011/PY/c1py00308a/c1py00308a-f9.gif) | ||
| Fig. 9 Effect of temperature on the degradation of Rh B in aqueous solution. [Rh B] = 5 × 10−5 mol L−1, [ZnPc-PAM] = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4. | ||
For practical use in water purification, the stability of the catalyst is also an important factor to be considered. Recycling experiments were performed for the photodegradation of Rh B over the polymeric catalyst. In each run, a new fresh solution of Rh B was supplied for maintenance of its initial concentration at 5 × 10−5 mol L−1. At the end of each run, the thermo-responsive catalyst was separated by elevating the temperature of the reaction system and dried in vacuum without further treatment. The catalytic activity was nearly unaffected as shown in Fig. 10, though the degradation rates in the next 5 reaction runs were slightly decreased which may be caused by the loss of the photocatalyst from one run to another. The results above indicate that the polymeric catalyst has a high stability during the recycling experiments.
![Recycling experiments for degradation of Rh B aqueous solution. [Rh B] = 5 × 10−5 mol L−1, [ZnPc-PAM]0 = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4.](/image/article/2011/PY/c1py00308a/c1py00308a-f10.gif) | ||
| Fig. 10 Recycling experiments for degradation of Rh B aqueous solution. [Rh B] = 5 × 10−5 mol L−1, [ZnPc-PAM]0 = 2 × 10−5 mol L−1, [H2O2] = 40 mL L−1, pH = 4. | ||
4. Conclusions
An end-functionalized PNIPAM with the asymmetrical phthalocyanine group, ZnPc-PAM, was synthesized viaATRP. The LCSTs of ZnPc-PAM with narrow polydispersity indices were higher than that of NIPAM homopolymer, which were attributed to the incorporation of the hydrophobic phthalocyanine group. After combination of phthalocyanine and PNIPAM, the well-defined ZnPc-PAM possessed the photocatalytic activity, verified by the experiment of the oxidative degradation of Rh B in aqueous solution. Due to the thermo-responsive property of ZnPc-PAM, it had the highest catalytic activity at around its LCST and could be recycled by the uncomplicated sedimentation through changing the temperature of the system.Notes and references
- S. Seelan, M. S. Agashe, D. Srinivas and S. Sivasanker, J. Mol. Catal. A: Chem., 2001, 168, 61–68 CrossRef CAS.
- S. Makhseed, F. Al-Kharafi, J. Samuel and B. Ateya, Catal. Commun., 2009, 10, 1284–1287 CrossRef CAS.
- J. F. van der Pol, E. Neeleman, R. J. M. Nolte, J. W. Zwikker and W. Drenth, Makromol. Chem., 1989, 190, 2727–2745 CrossRef CAS.
- V. Mantareva, V. Kussovski, I. Angelov, E. Borisova, L. Avramov, G. Schnurpfeil and D. Wöhrle, Bioorg. Med. Chem., 2007, 15, 4829–4835 CrossRef CAS.
- T. Matsuda, Y. Saito and K. Shoda, Biomacromolecules, 2007, 8, 2345–2349 CrossRef CAS.
- F. Yang, M. Shtein and S. R. Forrest, Nat. Mater., 2005, 4, 37–41 CrossRef CAS.
- G. Torre, P. Vázquez, F. Agulló-López and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef.
- B. Mukherjee and M. Mukherjee, Org. Electron., 2009, 10, 1282–1287 CrossRef CAS.
- N. Safari and F. Bahadoran, J. Mol. Catal. A: Chem., 2001, 171, 115–121 CrossRef CAS.
- K. P. Mishra and P. R. Gogate, Sep. Purif. Technol., 2010, 75, 385–391 CrossRef CAS.
- S. Mangematin and A. B. Sorokin, J. Porphyrins Phthalocyanines, 2001, 5, 674–680 CrossRef CAS.
- A. Shaabani and E. Farhangi, Appl. Catal., A, 2009, 371, 148–152 CrossRef CAS.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89, 6024–6030 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- I. Ankareddi and C. S. Brazel, Int. J. Pharm., 2007, 336, 241–247 CrossRef CAS.
- K. Fujimoto, C. Iwasaki, C. Arai, M. Kuwako and E. Yasugi, Biomacromolecules, 2000, 1, 515–518 CrossRef CAS.
- X. Z. Zhang, R. X. Zhuo, J. Z. Cui and J. T. Zhang, Int. J. Pharm., 2002, 235, 43–50 CrossRef CAS.
- T. Matsuda, Y. Saito and K. Shoda, Biomacromolecules, 2007, 8, 2345–2349 CrossRef CAS.
- S. S. Pennadam, M. D. Lavigne, C. F. Dutta, K. Firman, D. Mernagh, D. C. Górecki and C. Alexander, J. Am. Chem. Soc., 2004, 126, 13208–13209 CrossRef CAS.
- X. J. Lu, L. F. Zhang, L. Z. Meng and Y. H. Liu, Polym. Bull., 2007, 59, 195–206 CrossRef CAS.
- W. X. Chen, B. Y. Zhao, Y. Pan, Y. Y. Yao, S. S. Lu, S. L. Chen and L. J. Du, J. Colloid Interface Sci., 2006, 300, 626–632 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS.
- M. Kimura, H. Ueki, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Chem.–Eur. J., 2004, 10, 4954–4959 CrossRef CAS.
- Q. Duan, A. Narumi, Y. Miura, X. D. Shen, S. I. Sato, T. Satohand and T. Kakuchi, Polym. J., 2006, 38, 306–310 CrossRef CAS.
- Q. Duan, Y. Miura, A. Narumi, X. D. Shen, S. I. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1117–1124 CrossRef CAS.
- X. D. Tao, Z. G. Gao, T. Satoh, Y. Cui, T. Kakuchi and Q. Duan, Polym. Chem., 2011, 2, 2068 RSC.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
- J. Sleven, C. G. Walrand and K. Binnemans, Mater. Sci. Eng., C, 2001, 18, 229–238 CrossRef.
- M. Kimura, T. Kuroda, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Langmuir, 2003, 19, 4825–4830 CrossRef CAS.
- G. Masci, L. Giacomelli and V. Crescenzi, Macromol. Rapid Commun., 2004, 25, 559–564 CrossRef CAS.
- Y. Xia, X. C. Yin, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- Q. Zhang, B. He, Q. Dai, J. H. Gu, N. Gu and D. Y. Huang, Supramol. Sci., 1998, 5, 631–634 CrossRef CAS.
- B. M. Hassan, H. Li and N. B. McKeown, J. Mater. Chem., 2000, 10, 39–45 RSC.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- X. Tao, W. H. Ma, T. Y. Zhang and J. C. Zhao, Chem.–Eur. J., 2002, 8, 1321–1326 CrossRef CAS.
- Y. Fang and D. Y. Chen, Mater. Res. Bull., 2010, 45, 1728–1731 CrossRef CAS.
- L. Wu, A. Li, G. D. Gao, Z. H. Fei, S. R. Xu and Q. X. Zhang, J. Mol. Catal. A: Chem., 2007, 269, 183–189 CrossRef CAS.
- W. X. Chen, W. Y. Lü, X. Y. Shen and Y. Y. Yao, Sci. China Chem., 2010, 53, 638–644 CrossRef CAS.
- N. Barka, S. Qourzal, A. Assabbane, A. Nounah and Y. A. Ichou, J. Photochem. Photobiol., A, 2008, 195, 346–351 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |