DOI:
10.1039/C1PY00331C
(Paper)
Polym. Chem., 2011,
2, 2842-2849
Two-dimensional copolymers with D–A type side chains for organic thin-film transistors: Synthesis and properties†
Received
22nd July 2011
, Accepted 2nd September 2011
First published on 7th October 2011
Abstract
This paper reports the synthesis, characterization and field-effect transistor (FET) properties of two new two-dimensional (2D) polymers, P1 and P2. The conjugated polymers were constructed by electron-rich main chains and D–A type side chains. DSC and XRD measurements revealed the amorphous nature of the polymer thin films and electrochemistry measurement results showed a low HOMO level of about −5.1 eV for both polymers. The polymers based solution-processed thin-film transistors with bottom-contact/bottom-gate geometry were fabricated with the films annealed at different temperatures to evaluate the FET performances. The hole mobilities were 3.6 × 10−3 and 4.0 × 10−3 cm2 V−1s−1, with current on/off ratios of 1.0 × 105 and 2.0 × 105, for P1 and P2, respectively, at an annealing temperature of 160 °C when measured under ambient conditions, which are relatively good results for amorphous polymers.
Introduction
Organic semiconductors based on π-conjugated polymers have been widely studied in electronic applications.1–5 These polymers showed great advantages compared to their inorganic counterparts. Polymers based semiconductors can be solution processed, resulting in low-cost, large-area, and flexible active matrix display backplanes.6–13 Also, the polymers can be molecularly engineered to have tunable optical and electronic properties through synthesis and assembly,14,15 which can make them suit for various requirements in different electro-optical devices such as organic light emitting diodes (OLEDs), organic photovoltaic devices (OPVs), and organic thin-film transistors (OTFTs).
OTFTs have attracted intense interests in recent years, and significant progress has been achieved.16–25 To obtain high mobilities, the polymer semiconductors must be well ordered when processed from solution. Because polythiophene can exhibit a fine conjugated and coplanar conformation, and thus achieve a highly crystalline thin-film microstructure, they have provided amongst the best FET performance,26–31 with mobilities above 0.1 cm2 V−1s−1 and current on/off ratio above 106. Nonetheless, their stability still must be improved for practical applications.32 An effective approach to improve the stability and maintain the high mobility is to introduce electron-deficient aromatic rings into the backbone to shape donor–acceptor (D–A) copolymers.33–37 However, all these linear polymers, constructed by electron-rich thiophene derivatives or D–A segments, usually need careful processing to form a well aligned conformation in thin films. Therefore, 2D polymers with conjugated side chains,38–42 which have a larger conjugation face and provide more chances for molecular packing, may be a good choice for OTFTs. There may be no need for a strict process during the film formation to achieve high mobilities.
Benzodithiophene (BDT) as an electron-rich fused aromatic ring, which exhibits good planarity, is often introduced into the backbone of polymers to obtain high hole-mobility materials.43 On the other hand, benzothiazole as an electron-deficient unit could be used as an acceptor to shape the D–A structure. Herein, we designed and synthesized two new 2D polymers with electron-rich main chains and D–A type side chains, where BDT and various thiophene groups make up the main chains and benzothiazoles are connected to the main chains through a styryl unit to form the side chains (as shown in Fig. 1). Although the FET performances of some polymers with electron-rich side chains have been reported,44–46 there were few reports on 2D polymers with conjugated D–A type side chains for application on OTFTs. In this paper, we report the synthesis, thermal, optical, electrochemical, and FET properties of the polymers. The hole mobilities of the polymers reached the 10−3 cm2 V−1s−1 range with a current on/off ratio of 105 tested with bottom-contact/bottom-gate TFT devices under ambient conditions.
 |
| | Fig. 1 Chemical structures of the polymers. | |
Experimental
General information
1H NMR and 13C NMR spectra were measured on a MERCURY-VX300 spectrometer. Elemental analysis of carbon, hydrogen, and nitrogen was performed on a Vario EL III microanalyzer. UV-vis absorption spectra were recorded on a Shimadzu UV-2500 recording spectrophotometer. Gel permeation chromatography (GPC) analysis was performed on an Agilent 1100 series HPLC system equipped with a G1326A refractive index detector, in which polystyrene standards were used as calibration standards and THF was used as an eluent, and the flow rate was 1.0 mL min−1. Thermogravimetric analysis (TGA) was undertaken with a NETZSCH STA 449C instrument. The thermal stability of the samples under a nitrogen atmosphere was determined by measuring their weight loss while heating at a rate of 10 °C min−1. Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC822e unit at a heating rate of 10 °C min−1 from room temperature to 270 °C under nitrogen. Cyclic voltammetry (CV) was carried out in nitrogen purged anhydrous CH3CN solution at room temperature with a CHI voltammetric analyzer. Tetrabutylammonium hexafluorophosphate (TBAPF6) (0.1 M) was used as the supporting electrolyte. The conventional three-electrode configuration consisted of a platinum working electrode, a platinum wire auxiliary electrode, and an Ag wire pseudo-reference electrode with ferrocenium-ferrocene (Fc+/Fc) as the internal standard. Cyclic voltammograms were obtained at a scan rate of 100 mV s−1. Formal potentials were calculated as the average of cyclic voltammetric anodic and cathodic peaks. The onset potential was determined from the intersection of two tangents drawn at the rising and background current of the cyclic voltammogram. X-ray diffraction (XRD) was performed using a Rigaku-D/max-2500 X-ray diffractometer. Atomic force microscopy (AFM) images were obtained on a Nanoscope V AFM (Digital Instruments) in tapping mode.
Device fabrication and characterization
FET devices were fabricated with a bottom-gate, bottom-contact configuration. A heavily doped n-type Si wafer with a SiO2 layer of 300 nm and a capacitance of 11 nF cm−2 was used as the gate dielectric. Octyltrichlorosilane (OTS) was used as a self-assembled surface modifier for SiO2. The Au source/drain electrodes were prepared by photolithography. For spin-coated thin film device, a 50 nm-thick (±10 nm) semiconductor film was spin-coated on top of the OTS-treated SiO2 from 10 mg mL−1CHCl3 solution of the polymers. The channel length was 5 μm, and the channel width was 1400 μm. Device annealing was carried out at various temperatures for 1 h in a vacuum oven under a pressure of 0.1 Pa. The current–voltage (I–V) characteristics were measured with the Micromanipulator 6150 probe station in a clean and metallic shielded box at room temperature in air, and recorded with a Keithley 4200 SCS. The field-effect mobility was calculated in the saturation regime by using the equation IDS = (μWCi/2L)(VG − VT)2,18 where IDS is the drain-source current, μ is the field-effect mobility, W is the channel width, L is the channel length, Ci is the capacitance per unit area of the gate dielectric layer, VG is the gate voltage, and VT is the threshold voltage.
Materials
Toluene and THF were dried over and distilled from K-Na alloy under an atmosphere of argon. Compound 1, 6, and 8 was purchased from J&K Scientific Ltd. and used without further purification. Compound 2, 4, 7, 9 and 11 were synthesized according to literature procedures.47–51 Other reagents were obtained from Sinopharm Chemical Reagent Co. (Shanghai, China).
Synthesis of monomers
2-(p-Tolyl)benzo[d]thiazole (3).
Compound 2 (6.80 g, 44 mmol) was added slowly to the solution of compound 1 (5.51g, 44 mmol) in N-methyl-2-pyrrolidone (NMP, 100 mL) at room temperature, and then the mixture was heated at 110 °C for 1 h under argon. After cooling, the solution was poured into ice water, filtered and washed with cold ethanol. The solid was collected and purified by recrystallization from ethanol. Yield: 7.9 g, 80%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.05 (d, J = 7.8 Hz, 1H), 7.98 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.1 Hz, 2H), 7.48 (m, 1H), 7.37 (m, 1H), 7.30 (d, J = 7.2 Hz, 2H), 2.42 (s, 3H).
2-(4-(Bromomethyl)phenyl)benzo[d]thiazole (4).
Compound 3 (4.05 g, 18 mmol) was dissolved in 40 mL of CCl4, then benzoyl peroxide (BPO, 45 mg, 1% mmol) and NBS (3.60 g, 20.2 mmol) was added to the solution at once. The mixture was refluxed for 12 h. After cooling, the solution was filtered and the filtrate was distilled under reduced pressure to get the crude product, then it was recrystallized form ethyl acetate to get pure white solid. Yield: 4.66 g, 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.11–7.89 (m, 4H), 7.53–7.37 (m, 4H), 4.53 (s, 2H)
Diethyl 4-(benzo[d]thiazol-2-yl)benzylphosphonate (5).
A mixture of 2-(4-(bromomethyl)phenyl)benzo[d]thiazole (4, 1 g, 3.29 mmol) and 5 ml of triethyl phosphite was heated at 120 °C for 16 h. Excess triethyl phosphite was removed by vacuum distillation. The crude product was purified by column chromatography on silica gel (CHCl3). Yield: 1 g, 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.09–8.04 (m, 3H), 7.92 (d, J = 8.1 Hz, 1H), 7.53–7.35 (m, 4H), 4.10–4.00 (m, 4H), 3.23 (d, J = 22 Hz, 2H), 1.27 (t, J = 6.9 Hz, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 168.3, 153.3, 135.8, 134.7, 131.8, 130.7, 128.1, 126.8, 125.7, 123.1, 121.8, 62.5, 34.6, 33.7, 16.6. Anal. Calcd for C18H20NO3PS: C, 59.82; H, 5.58; N, 3.88. Found: C, 59.89; H, 5.42; N, 3.87.
2,5-Dibromothiophene-3-carbaldehyde (7).
To a solution of compound 6 (2.50 g, 22.3 mmol) in 100 ml of CHCl3 was added anhydrous NaHCO3 (4.4 g, 52.3 mmol), followed by the dropwise addition of a solution of bromine (8.2 g, 51.2 mmol, in 50 mL of chloroform) over a period of 1 h. The reaction mixture was stirred overnight at room temperature and then filtered. The filtrate was washed with saturated Na2SO3 solution and dried over MgSO4. The solvent was evaporated, and the solid residue was purified by column chromatography on silica gel with petroleum ether–ethyl acetate (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as eluent. Yield: 5.21 g, 86%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 9.79 (s, 1H), 7.34 (s, 1H).
:1, v/v) as eluent. Yield: 5.21 g, 86%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 9.79 (s, 1H), 7.34 (s, 1H).
(E)-2-(4-(2-(2,5-Dibromothiophen-3-yl)vinyl)phenyl)benzo[d]thiazole (M1).
Compound 5 (0.5 g, 1.39 mmol) and 2,5-dibromothiophene-3-carbaldehyde (7) (0.37 g, 1.39 mmol) were dissolved in 20 mL of anhydrous DMF. To this solution was added NaH (0.15 g, 6.25 mmol) in an ice-water bath. Then the reaction was stirred for 5 h at room temperature under argon. After the reaction, the mixture was poured into water. The precipitate was filtered and washed with water and methanol. The crude product was purified by column chromatography (petroleum ether–CHCl3, 1:1). Yield: 0.46 g, 70%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.10–8.07 (m, 3H), 7.92 (d, J = 7.8 Hz, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.51 (t, J = 7.5 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 3.3 Hz, 1H), 7.11 (d, J = 16.2 Hz, 1H), 6.94 (d, J = 16.2 Hz, 1H). 13C NMR (CDCl3, 150 MHz) δ [ppm]: 139.79, 131.72, 131.46, 131.14, 131.09, 128.68, 128.44, 128.07, 127.80, 127.65, 126.31, 125.72, 125.44, 125.37, 125.26, 125.09, 123.80, 122.97, 122.89, 112.26, 110.66. Anal. calcd for C19H11Br2NS2: C, 47.82; H, 2.32; N, 2.93. Found: C, 48.09; H, 2.23; N, 3.04.
2-(4-Dodecylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (9).
To a solution of 3-dodecylthiophene (8, 5.0 g, 19.8 mmol) in THF (70 mL) at 0 °C, 10 mL of LDA (2 M in hexane) was added by syringe and the mixture was stirred for 1 h. Then the mixture was cooled to −78 °C, and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4.5 mL, 22.1 mmol) was added to the solution. After 1 h, the solution was warmed to room temperature, and stirred for a further 12 h. The mixture was poured into water, extracted with diethyl ether, and dried over anhydrous Na2SO4. The solvent was removed viarotary evaporation, and the resulting oil was purified by column chromatography on silica gel using ethyl acetate–petroleum ether (1:100) as eluent. Yield: 6.03 g, 80%. 1H NMR (300 MHz, CDCl3, δ): 7.47 (s, 1H), 7.21 (s, 1H), 2.62 (t, J = 8.1 Hz, 2H), 1.61 (m, 2H), 1.34–1.25 (m, 30H), 0.88 (t, J = 6 Hz, 3H).
Compound 10.
M1 (0.414 g, 0.87 mmol), compound 9 (0.72 g, 1.9 mmol) and Pd (PPh3)4 (66 mg, 3% mol) were mixed in a Schlenk tube filled with argon. Then K2CO3 solution (2 M aq. 4 mL) and THF (12 mL) were injected into the reactor through a syringe. The mixture was stirred at 70 °C for 48 h. After the reaction, the resulting mixture was cooled to room temperature and then poured into water and extracted with ethyl ether twice. The organic extracts were collected and dried with anhydrous Na2SO4. After removal of the solvent, the crude product was purified by column chromatography on silica gel using petroleum ether–chloroform (10:1) as eluent. Yield: 0.6 g, 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.10 (d, J = 7.2 Hz, 3H), 7.90 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 2.7 Hz, 1H), 7.46 (s, 1H), 7.42–7.39 (m, 2H), 7.07–7.00 (m, 4H), 6.85 (s, 1H), 2.62 (m, 4H), 1.67–1.65 (m, 4H), 1.33–1.27(m, 36H), 0.88–0.87 (m, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 167.6, 154.1, 144.2, 144.1, 140.2, 136.3, 135.7, 135.0, 134.7, 132.9, 132.4, 129.0, 128.4, 127.9, 126.9, 126.3, 125.5, 125.1, 123.5, 123.1, 121.7, 121.5, 121.3, 119.5, 31.91, 30.50, 30.46, 30.37, 29.66, 29.51, 29.47, 29.35, 22.68, 14.11. Anal. calcd for C51H65NS4: C, 74.67; H, 7.99; N, 1.71. Found: C, 74.86; H, 7.88; N, 1.38.
Synthesis of M2.
Compound 10 (0.43 g, 0.52 mmol) was dissolved in a mixture of chloroform (30 mL) and glacial acetic acid (30 mL). NBS (0.20 g, 1.12 mmol) was then added to the solution and stirred for 12 h in the dark. Much precipitate appeared during the reaction. After the reaction, the mixture was poured into water, and extracted with chloroform. The organic extracts were collected and dried with anhydrous Na2SO4. After removal of the solvent, the crude product was purified by column chromatography on silica gel using petroleum ether/chloroform (1:3) as eluent. Pure M2 as a yellow powder was obtained. Yield: 0.46 g, 90%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.11–8.07 (m, 3H), 7.91 (d, J = 8.1 Hz, 1H), 7.59(d, J = 8.7 Hz, 2H), 7.53 (t, J = 8.1 Hz, 1H), 7.42–7.32 (m, 3H), 7.04 (d, J = 16.2 Hz, 1H), 6.90 (d, J = 6 Hz, 2H), 2.63–2.53 (m, 4H), 1.63–1.61 (m, 4H), 1.33–1.27 (m, 36H), 0.87 (t, J = 5.9 Hz, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 167.7, 154.4, 143.4, 143.3, 140.0, 136.5, 136.1, 135.9, 135.3, 134.6, 133.0, 132.0, 130.0, 128.1, 127.2, 126.5, 125.4, 125.2, 123.4, 122.8, 122.1, 121.8, 110.5, 108.7, 32.17, 29.94, 29.91, 29.86, 29.83, 29.70, 29.66, 29.60, 29.51, 22.93, 14.26. Anal. calcd for C51H63Br2NS4: C, 62.62; H, 6.49; N, 1.43. Found: C, 62.87; H, 6.31; N, 1.63.
Synthesis of P1.
Compound 11 (0.2825 g, 0.3193 mmol), M1 (0.1524g, 0.3195 mmol) and Pd (PPh3)4 (8 mg, 2% mol) were dissolved in toluene (12 mL). The polymerization was carried out at 110 °C for 3 days under argon. After the reaction, the mixture was cooled to room temperature and the polymer was precipitated by slowly adding the mixture into CH3OH (200 mL). The precipitated material was collected by filtration through a funnel. Then the crude product was extracted via a Soxhlet apparatus with methanol, acetone, hexane, and finally chloroform. Only the chloroform-soluble portion was collected, concentrated, and precipitated from methanol to yield P1 as a red solid. Yield: 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.11–6.91 (m, 13H), 4.32 (br, 4H), 1.58–0.85 (m, 46H). GPC: Mn = 36.9 k, Mw/Mn = 2.97.
Synthesis of P2.
Compound 11 (0.1232 g, 0.1393 mmol), M2 (0.1362 g, 0.1392 mmol) and Pd (PPh3)4 (4 mg, 2% mol) were dissolved in toluene (12 mL). The polymerization was carried out at 110 °C for 3 days under argon. After the reaction, the mixture was cooled to room temperature and the polymer was precipitated by slowly adding the mixture into CH3OH (200 mL). The precipitated material was collected by filtration through a funnel. Then the crude product was extracted via a Soxhlet apparatus with methanol, acetone, hexane, and finally chloroform. Only the chloroform-soluble portion was collected, concentrated, and precipitated from methanol to yield P2 as a red solid. Yield: 75%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.11 (br, 4H), 7.88 (br, 2H), 7.63–7.39 (m, 7H), 7.10 (br, 2H), 4.32 (br, 4H), 2.92 (br, 4H), 1.94 (br, 4H), 1.78 (br, 8H), 1.62 (br, 4H), 1.26 (br, 64H), 0.86 (br, 12H). GPC: Mn = 15.9 k, Mw/Mn = 3.07.
Results and discussion
Synthesis
The synthetic routes of M1 and M2 are shown in Scheme 1. P1 and P2 were synthesized by a Stille coupling reaction as shown in Scheme 2. After the polymerization reaction was completed, the crude polymers were precipitated from methanol and extracted via a Soxhlet apparatus with methanol, acetone, hexane, and finally chloroform. Only the chloroform-soluble portion was collected in order to obtain a pure, high molecular weight, solution-processable polymer. Both polymers have good solubility in common organic solvents such as toluene, chloroform, THF and chlorobenzene. Molecular weights and polydispersity indices (PDIs) of polymers, as shown in Table 1, are determined by GPC analysis in THF with a polystyrene standard calibration.
 |
| | Scheme 1 Synthetic routes of the monomers. Conditions: (a) NMP, 110 °C; (b) NBS, BPO, CCl4, reflux; (c) P(OEt)3, 120 °C; (d) Br2, NaHCO3, CHCl3; (e) NaH, DMF; (f) LDA, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF; (g) Pd(PPh3)4, K2CO3 (2M, aq), THF; (h) NBS, CHCl3–CH3COOH. | |
 |
| | Scheme 2 Synthetic routes of P1 and P2. | |
Table 1 Molecular weights and thermal properties of the polymers
|
Polymers
|
M
n/g mol−1 |
M
w/g mol−1 |
PDI (Mw/Mn) |
T
d/°C |
|
P1
|
3.69 × 104 |
1.10 × 105 |
2.97 |
333.5 |
|
P2
|
1.59 × 104 |
4.88 × 104 |
3.07 |
343.2 |
Thermal properties
The thermal properties of the polymers were evaluated by TGA and DSC. As shown in Fig. 2, excellent thermal stability of the polymers were manifested in its TGA profile showing a decomposition temperature of 333.5 and 343.2 °C, respectively (5% weight loss), which is high enough for device fabrication. No thermal transitions were detected within the range of the study (25–270 °C) in our DSC results, indicating the amorphous nature of the polymers.
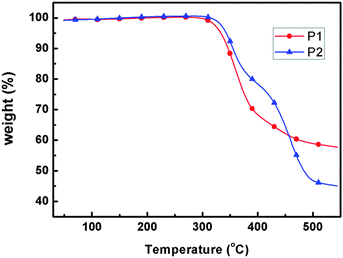 |
| | Fig. 2
TGA plots of the polymers with a heating rate of 10 °C min−1 under nitrogen atmosphere. | |
Table 2 Optical and electrochemical data of the polymers
|
Polymer
|
UV-vis absorption |
Cyclic voltammetry
|
|
CHCl3 solutiona |
Filmb |
|
λ
max/nm |
λ
onset/nm |
E
g/eVc |
λ
max/nm |
λ
onset/nm |
E
g/eVc |
E
oxonset/V; HOMO/eV |
E
redonset/V; LUMO/eV |
|
Measured in chloroform solution.
Cast from chloroform solution.
Band gaps estimated from the onset wavelength (λedge) of the optical absorption: Eg = 1240/λedge.
|
|
P1
|
361 |
520 |
622 |
1.99 |
366 |
536 |
632 |
1.96 |
0.28; −5.08 |
−2.06; −2.74 |
| P2 |
363 |
463 |
562 |
2.21 |
367 |
531 |
647 |
1.92 |
0.31; −5.11 |
−2.12; −2.68 |
Electrochemical properties
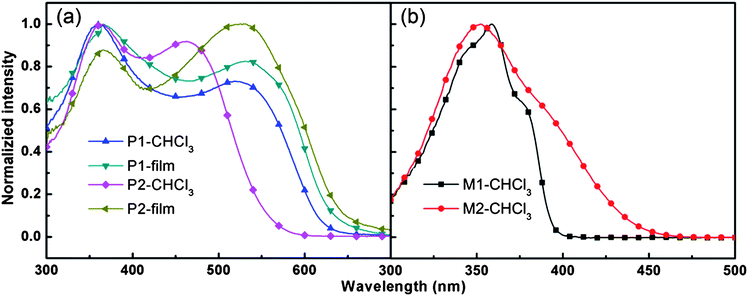
Electrochemical property is important for conjugated polymers, upon which many applications of the polymers depend. And CV was widely employed to test the electrochemical properties, estimating the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the polymers. Fig. 4 shows the cyclic voltammograms of the two polymer films on Pt disk electrode in 0.1 mol L−1Bu4NPF6 acetonitrile solution. The HOMO and LUMO energy levels were calculated from the onset oxidation potentials and onset reduction potentials.52–54 The energy band gaps of P1 and P2 were 2.34 eV and 2.43 eV, respectively, significantly larger than their optical band gaps (around 1.95 eV). The discrepancy might have been caused by the presence of an energy barrier at the interface between the polymer film and the electrode surface.55–57 The HOMO levels of both polymers are around −5.1 eV, which is lower than that of P3HT (−4.9 eV), therefore, it should show higher stability in device fabrication. In addition, the HOMO level will match the work function of gold and ensure effective hole injection between a gold electrode and the semiconducting layer.
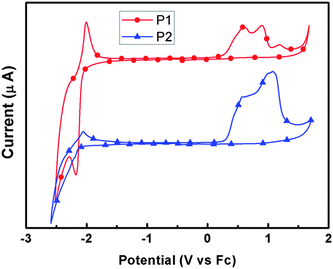 |
| | Fig. 4
Cyclic voltammograms of P1 and P2 films on a platinum electrode measured in 0.1 mol L−1Bu4NPF6 acetonitrile solutions at a scan rate of 100 mV s−1. | |
Molecular orbital calculations
The geometries of oligomer structures of the two copolymers were optimized at the Density Functional Theory (DFT) ri-BP86/def2-SVP level, then the electronic properties were calculated at B3LYP/6–31g(d) level.58,59 The frontier energy levels (HOMO and LUMO) were characterized for the neutral electronic states. Note that the calculations were performed on model systems of copolymers where all alkyl chain substituents were replaced with methyl groups (this has only a minimal effect on the electronic properties).
As shown in Fig. 5, the LUMO levels of the polymers are delocalized over the whole molecules, indicating that the side chain groups are conjugated to the main chains very well. On the other hand, the HOMO levels are mainly delocalized along the main chains and partially extend to the side chains, which may be due to the electron-deficient property of the benzothiazole unit at the end of the side chains. These results confirmed that P1 and P2 showed good conjugation properties, which would be beneficial to charge transport. Moreover, the delocalization of HOMO and LUMO levels along the main chains agrees well with the idea that the longer wavelength absorptions in the UV-vis spectra come from the π–π* transitions of the polymers.
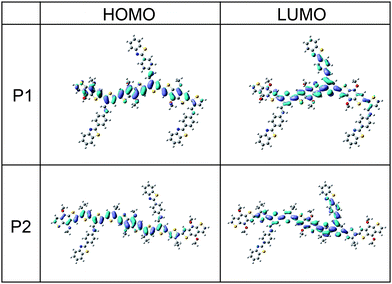 |
| | Fig. 5 HOMO and LUMO wavefunctions of the two oligomer (n = 3 for P1 and n = 2 for P2) model systems calculated at the B3LPY/6–31g(d) level. | |
Film morphology
The film morphology of the polymers was examined using AFM. Fig. 6 shows the tapping-mode AFM height images of spin-coated P1 and P2 films on an OTS-modified SiO2 substrate after annealing at 160 °C. P2 showed a more uniform surface than P1, but no obvious crystalline areas were found in either image. To further understand the AFM results, structural ordering of the molecules in thin films was evaluated using XRD. And the XRD analysis confirmed the amorphous nature of the films as no Bragg refraction peaks were detected even after thermal annealing at 160 °C, which also agrees with the DSC results.
 |
| | Fig. 6
AFM height images of the polymer films annealed at 160 °C (a) P1 and (b) P2. | |
FET properties
Bottom-contact/bottom-gate TFTs were fabricated from the two polymer materials by solution-processing techniques. OTS-treated Si–SiO2 substrates were chosen and the polymers were annealed at different temperatures in vacuum in order to adjust the order of the thin-films and to find the best conditions for device fabrication (ESI†). Fig. 7 shows the typical output and transfer curves of P1 and P2 at a representative annealing temperature of 160 °C as p-channel FETs. The output behaviors of P2 followed closely the gradual-channel model employed in metal oxide semiconductor FETs with very good saturation and no observable contact resistance. The transfer characteristics showed small threshold voltages for both polymer-based devices. As shown in Table 3, when processed at room temperature, a thin film of P1 showed a hole mobility of 1.2 × 10−3 cm2 V−1s−1, and as the annealing temperature increased, the mobility improved steadily. The highest mobility reached 3.6 × 10−3 cm2 V−1s−1, which is 3 times larger than that before annealing, and a good on/off ratio of current was obtained as high as 1.0 × 105 when the annealing temperature was 160 °C. The same tendency was also found for P2. The best performances are also reached after annealing at 160 °C, with a hole mobility of 4.0 × 10−3 cm2 V−1s−1 that is enhanced 6 times versus that before annealing, and with an Ion/Ioff of 2.0 × 105. From the data above, it was revealed that P2 showed more improvement in its FET properties when annealing processes were applied in device fabrication. This can be explained from the design rule of the polymers. We can see that 2 more 3-dodecylthiophene units were introduced into the backbone of P2, which could make the polymer main chain more extended with less steric hindrance. Therefore, the segments of P2 would have more freedom to rotate and then could be more easily packed close to each other so as to be more sensitive to temperature.
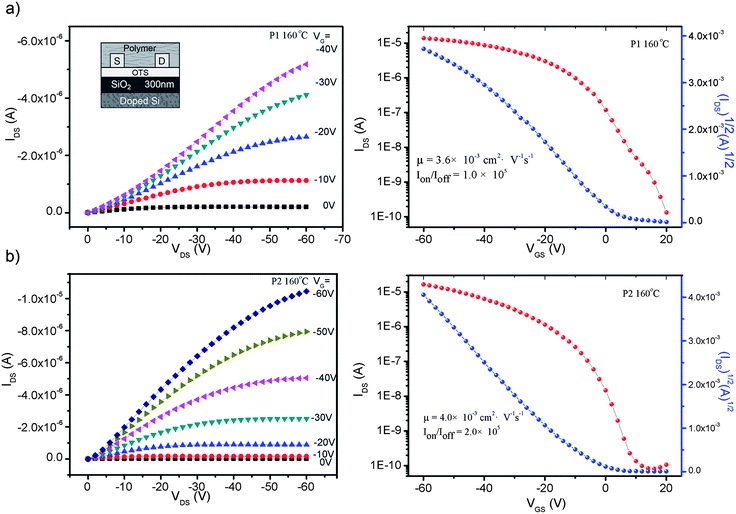 |
| | Fig. 7 Output characteristics at different gate bias (left) and transfer characteristics at constant source–drain voltage, VD = −60 V (right) for devices using (a) P1 and (b) P2 as semiconductors when annealed at 160 °C in vacuum. | |
Table 3 The hole mobility, on/off current ratio and threshold voltage of solution-processed TFTs based on P1 and P2 when annealed at different temperatures
|
T/°C |
P1
|
P2 |
|
μ/cm2 V−1s−1 |
I
on/Ioff |
V
T/V |
μ/cm2 V−1s−1) |
I
on/Ioff |
V
T/V |
|
R.T. |
1.2 × 10−3 |
8.3 × 102 |
22 |
6.2 × 10−4 |
7 × 103 |
21.2 |
| 80 |
1.78 × 10−3 |
1.6 × 104 |
17.9 |
1.2 × 10−3 |
4 × 104 |
13.5 |
| 120 |
3.0 × 10−3 |
5.0 × 104 |
16.6 |
2.5 × 10−3 |
1.4 × 105 |
10.1 |
| 160 |
3.6 × 10−3 |
1.0 × 105 |
13.1 |
4.0 × 10−3 |
2.0 × 105 |
7.7 |
Conclusion
We have successfully synthesized two new 2D conjugated polymers with D–A type side chains. The polymers are soluble in common solvents and have good thermal stability. DSC and XRD analysis showed that the polymer thin films were amorphous. The CV measurements revealed that they are typical semiconductors applied for TFTs. Bottom-contact/bottom-gate TFTs were fabricated with solution-processed polymer films. The FET properties of the polymers were tested under ambient conditions with the films annealed at different temperatures, and the best performance was obtained with a hole mobility of 3.6 × 10−3 and 4.0 × 10−3 cm2 V−1s−1, and current on/off ratios of 1.0 × 105 and 2.0 × 105, for P1 and P2, respectively, at the annealing temperature of 160 °C, which are relatively good results for amorphous polymers.
Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 20772094).
References and notes
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS.
- P. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- Y. Cheng, S. Yang and C. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- J. Ohshita, Macromol. Chem. Phys., 2009, 210, 1360–1370 CrossRef CAS.
- M. T. Bernius, M. Inbasekaran, J. O'Brien and W. Wu, Adv. Mater., 2000, 12, 1737–1750 CrossRef CAS.
- F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, Science, 1994, 265, 1684–1686 CAS.
- H. E. Katz, Chem. Mater., 2004, 16, 4748–4756 CrossRef CAS.
- R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 222–228 CrossRef.
- H. Yang, T. J. Shin, L. Yang, K. Cho, C. Y. Ryu and Z. Bao, Adv. Funct. Mater., 2005, 15, 671–676 CrossRef CAS.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741–1744 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS.
- M. Mushrush, A. Facchetti, M. Lefenfeld, H. E. Katz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 9414–9423 CrossRef CAS.
- J. Liu, R. Zhang, G. Sauvé, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2008, 130, 13167–13176 CrossRef CAS.
- Z. Bao, J. A. Roger and H. E. Katz, J. Mater. Chem., 1999, 9, 1895–1904 RSC.
- J. Locklin, D. Li, S. C. B. Mannsfeld, E. Borkent, H. Meng, R. Advincula and Z. Bao, Chem. Mater., 2005, 17, 3366–3374 CrossRef CAS.
- H. E. Katz, Z. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359–369 CrossRef CAS.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- H. Fukuda, M. Ise, T. Kogure and N. Takano, Thin Solid Films, 2004, 464–465, 441–444 CrossRef CAS.
- E. Lim, Y. M. Kim, J. Lee, B. Jung, N. S. Cho, J. Lee, L. Do and H. Shim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4709–4721 CrossRef CAS.
- R. A. Street, Adv. Mater., 2009, 21, 2007–2022 CrossRef CAS.
- T. L. Nelson, T. M. Young, J. Liu, S. P. Mishra, J. A. Belot, C. L. Balliet, A. E. Javier, T. Kowalewski and R. D. McCullough, Adv. Mater., 2010, 22, 4617–4621 CrossRef CAS.
- M. Tong, S. Cho, J. T. Rogers, K. Schmidt, B. B. Y. Hus, D. Moses, R. C. Coffin, E. J. Kramer, G. C. Banza and A. J. Heeger, Adv. Funct. Mater., 2010, 20, 3959–3965 CrossRef CAS.
- L. Zhang, C. Di, G. Yu and Y. Liu, J. Mater. Chem., 2010, 20, 7059–7073 RSC.
- Y. Zhao, C. Di, X. Gao, Y. Hu, Y. Guo, L. Zhang, Y. Liu, J. Wang, W. Hu and D. Zhu, Adv. Mater., 2011, 23, 2448–2453 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS.
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, Adv. Mater., 2005, 17, 1141–1144 CrossRef CAS.
- I. Mcculloch, M. Heeney, C. Bailey, K. Genevicius, I. Macdonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. Mcgehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. DeLongchamp, R. J. Kline, M. Cölle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091–1109 CrossRef CAS.
- K. H. Kim, D. S. Chung, C. E. Park and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 54–64 Search PubMed.
- H. Sirringhaus, Adv. Mater., 2009, 21, 3859–3873 CrossRef CAS.
- X. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206–7207 CrossRef CAS.
- Y. Li, S. P. Singh and P. Sonar, Adv. Mater., 2010, 22, 4862–4866 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C. Liu, S. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS.
- J. H. Kwon, J. An, H. Jang, S. Choi, D. S. Chung, M. Lee, H. Cha, J. Park, C. Park and Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1119–1128 CrossRef CAS.
- F. Huang, K. Chen, H. Yip, S. K. Hau, O. Acton, Y. Zhang, J. Luo and A. K.-Y. Jen, J. Am. Chem. Soc., 2009, 131, 13886–13887 CrossRef CAS.
- Y. Chang, S. Hsu, M. Su and K. Wei, Adv. Mater., 2009, 21, 2093–2097 CrossRef CAS.
- J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS.
- H. Fan, Z. Zhang, Y. Li and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1462–1470 CrossRef CAS.
- D. Chen, Y. Yang, C. Zhong, Z. Yi, F. Wu, L. Qu, Y. Li, Y. Li and J. Qin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3852–3862 CrossRef CAS.
- Y. Liang, Z. Xu, J. Xia, S. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS.
- Y. Zou, W. Hu, G. Sang, Y. Yang, Y. Liu and Y. Li, Macromolecules, 2007, 40, 7231–7237 CrossRef CAS.
- Y. Zou, G. Sang, W. Hu, Y. Liu and Y. Li, Synth. Met., 2009, 159, 182–187 CrossRef CAS.
- B. Liu, W. Wu, B. Peng, Y. Liu, Y. He, C. Pan and Y. Zou, Polym. Chem., 2010, 1, 678–684 RSC.
- S. S. Chaudhari and K. G. Akamanchi, Synlett, 1999, 1763–1765 CrossRef CAS.
- T. Liu, G. S. He, P. N. Prasad and L. Tan, J. Mater. Chem., 2004, 14, 982–991 RSC.
- Y. Zhang, C. B. Murphy and W. E. Jones, Macromolecules, 2002, 35, 630–636 CrossRef CAS.
- H. Kong, D. S. Chung, I. Kang, J. Park, M. Park, I. H. Jung, C. E. Park and H. Shim, J. Mater. Chem., 2009, 19, 3490–3499 RSC.
- J. Hou, T. L. Chen, S. Zhang, H. Chen and Y. Yang, J. Phys. Chem. C, 2009, 113, 1601–1605 CAS.
- J. L. Bredas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef CAS.
- D. W. De Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bssler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- D. A. M. Egbe, L. H. Nguyen, H. Hoppe, D. Mühlbacher and N. S. Sariciftci, Macromol. Rapid Commun., 2005, 26, 1389–1394 CrossRef CAS.
- Z.-K. Chen, W. Huang, L.-H. Wang, E.-T. Kang, B. J. Chen, C. S. Lee and S. T. Lee, Macromolecules, 2000, 33, 9015–9025 CrossRef CAS.
- G. Chen, Y. Cheng, Y. Chou, M. Su, C. Chen and K. Wei, Chem. Commun., 2011, 47, 5064–5066 RSC.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
Footnote |
| † Electronic supplementary information (ESI) available: Output and transfer curves using P1 and P2 as semiconductors when annealed at different temperatures. See DOI: 10.1039/c1py00331c |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.