DOI:
10.1039/C1PY00327E
(Paper)
Polym. Chem., 2011,
2, 2900-2906
Downwards tuning the HOMO level of polythiophene by carboxylate substitution for high open-circuit-voltage polymer solar cells
Received
20th July 2011
, Accepted 8th October 2011
First published on 21st October 2011
Abstract
Three polythiophene derivatives with carboxylate substituents, PT–C1, PT–C2 and PT–C3 were synthesized by the Pd-catalyzed Stille-coupling method, for downwards tuning of the highest occupied molecular orbital (HOMO) of polythiophene. The polymers show similar visible absorption spectra to that of regioregular poly(3-hexylthiophene) (P3HT). By attaching the carboxylate substituent, the HOMO energy level downwards shifted to −5.15, −5.11 and −5.10 eV for PT–C1, PT–C2 and PT–C3, respectively, in comparison with that (ca. −4.8 eV) for P3HT. The polymer solar cells (PSCs) based on the three polymers as donor and PC70BM as acceptor all exhibit relatively high open circuit voltage (Voc) of ca. 0.8 V. The power conversion efficiency (PCE) of the PSCs based on PT–C3: PC70BM = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 (w/w) reached 3.87% with Voc = 0.78 V, short circuit current (Jsc) of 9.68 mA cm−2 and fill factor (FF) of 51.2% under the illumination of AM1.5, 100 mW cm−2. These results indicate that attaching the carboxylate substituent is a simple and effective way to modulate energy levels of polythiophene derivatives and PT–C3 is a promising donor material for PSCs.
:1.5 (w/w) reached 3.87% with Voc = 0.78 V, short circuit current (Jsc) of 9.68 mA cm−2 and fill factor (FF) of 51.2% under the illumination of AM1.5, 100 mW cm−2. These results indicate that attaching the carboxylate substituent is a simple and effective way to modulate energy levels of polythiophene derivatives and PT–C3 is a promising donor material for PSCs.
Introduction
In the past few years, polymer solar cells (PSCs) have attracted much attention because of their advantages of low cost, easy fabrication, light weight, and the capability to fabricate flexible large-area devices.1–5 Bulk heterojunction (BHJ) solar cells utilizing a conjugated polymer as donor and fullerene derivatives [6,6]-phenyl-C61-butyric acid methyl ester (PC60BM) or [6,6]-phenyl-C71-butyric acid methyl ester (PC70BM) as acceptor have been proven to be the most successful device structure. Among the active layer materials in PSCs, regioregular poly(3-hexylthiophene) (P3HT) is one of the most important photovoltaic donor materials and has attracted much attention. Although the photovoltaic performance of P3HT/PCBM-based PSC devices can reach up to 4–5%,6 the Voc of the devices is limited to ∼0.6 V due to the relatively higher-lying HOMO (ca. −4.8 eV7) of P3HT. Recently, our group synthesized a new acceptor indene-C60 bisadduct (ICBA) with 0.17 eV higher LUMO energy level than that of PCBM,8 and the PSCs based on P3HT/ICBA demonstrated a high Voc of 0.84 V and a higher PCE of 6.48%.9 On the other hand, improving Voc of the device by molecular structure design of polythiophene should also be an interesting issue in PSCs.
Incorporation of aromatic heterocyclic rings such as thiazolothiazole,10bithiazole,11pyridopyrazine,12phthalimide13 and thienopyrroledione14 into a polythiophene backbone by copolymerization represents a successful strategy, leading to polymer semiconductors with enhanced oxidative stability compared to the parent poly(3-alkylthiophene). Changing the substituent also has been proven to be an effective way to decrease the HOMO energy level of polymers. By reducing the number of electron donating groups (the alkyl side chains) in the polythiophene backbone, Hou et al. synthesized P3HDTTT with deep HOMO of −5.3 eV, without changing their optical band gap.15 The Voc of the P3HDTTT/PCBM based device reached 0.82 V, which is ∼0.2 V higher than that of the P3HT/PCBM-based device. Meanwhile, a new polymer P3CTV was synthesized by attaching the carboxylate substitution instead of the hexyl side chain of poly(3-hexylthienylene vinylene) (P3HTV).16 Compared with P3HTV, the HOMO level of P3CTV shifted downwards by 0.21 eV. The P3CTV/PCBM based device attained a relatively high Voc of 0.86 V.
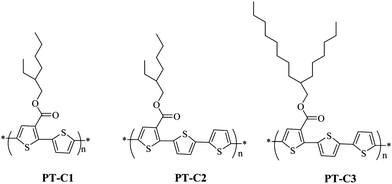
Here, we synthesized three polythiophene derivatives PT–C1, PT–C2 and PT–C3 (see Scheme 1), containing a carboxylate substituted group in the 3-position of thiophene by Stille coupling reaction. The chemical structure, molecular weight, thermal stability, electrochemical and photovoltaic properties of the polymers were characterized. Bulk heterojunction PSCs based on PT–C3: PC70BM = 1:1.5(w/w) show power conversion efficiencies up to 3.87% with Voc = 0.78V, short circuit current (Jsc) of 9.68 mA cm−2 and fill factor of 51.7% under the illumination of AM 1.5, 100 mW cm−2. It should be mentioned that during waiting for a patent application for this work, we found a publication17 on a polymer with same structure as PT–C3, but our polymer PT–C3 possesses much higher molecular weight and shows better photovoltaic performance.
 |
| | Scheme 1 Molecular structures of the polythiophene derivatives. | |
Results and discussion
Material synthesis
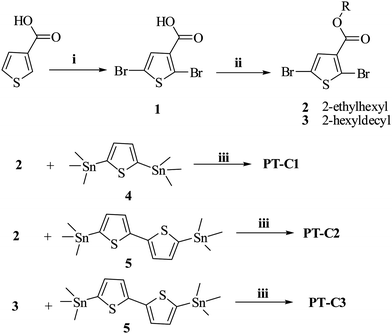
The general synthetic strategy for monomers and polymers is outlined in Scheme 2. The polymers of PT–C1, PT–C2, and PT–C3 were synthesized by Stille coupling reaction. All the polymers have good solubility in common organic solvents such as chloroform, toluene, chlorobenzene, dichlorobenzene, and so on. Molecular weights and polydispersity indices (PDIs) of the polymers, as listed in Table 1, are determined by gel permeation chromatography (GPC) analysis with a polystyrene standard calibration and THF as the eluent. The number average molecular weight (Mn) of PT–C3 reached 69.4 K, but the Mn values of PT–C1 and PT–C2 were only 4.3 K and 3.2 K respectively. The low molecule weights of PT–C1 and PT–C2 might be caused by the short alkyl side chain compared to PT–C3.
 |
| | Scheme 2 The synthesis routes for the monomers and polymers. Conditions: (i) Br2, acetic acid, 60 °C, reflux, 15 h; (ii) 2-ethylhexyl-1-ol or 2-hexyldecan-1-ol, DMAP, DCC, CH2Cl2, nitrogen, room temperature, 6 h; (iii) Pd(PPh3)4, toluene, refluxing. | |
Table 1 Molecular weights and thermal properties of the polymers
|
Polymers
|
M
w
a
|
M
n
a
|
PDIa |
T
d (°C)b |
|
M
n, Mw, and PDI of the polymers were determined by GPC using polystyrene standards in THF.
The 5% weight-loss temperatures under inert atmosphere.
|
|
PT
–C1
|
6.9K
|
4.3K
|
1.62 |
290 |
|
PT
–C2
|
5.6K
|
3.2K
|
1.75 |
355 |
|
PT
–C3
|
113.4K
|
69.4K
|
1.63 |
358 |
The thermal stability of the three polythiophene derivatives was measured by thermogravimetric analysis (TGA), as shown in Fig. 1. The TGA analysis reveals that the onset temperature with 5% weight-loss of PT–C1, PT–C2 and PT–C3 is 290, 355 and 358 °C, respectively (see Table 1). This indicates that the thermal stability of the three polythiophene derivatives is good enough for application in optoelectronic devices.
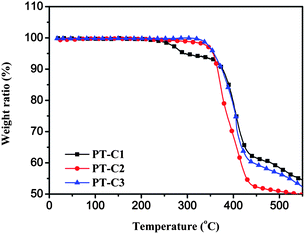 |
| | Fig. 1
TGA plots of the polymers with a heating rate of 10 °C min−1 under inert atmosphere. | |
Optical properties
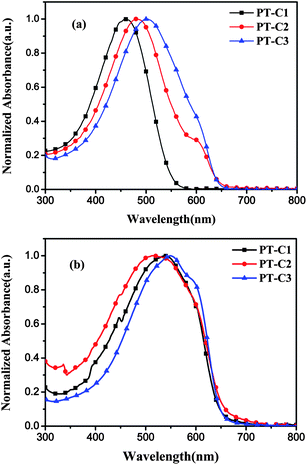
The photophysical characteristics of the polymers were investigated by ultraviolet–visible (UV–vis) absorption spectroscopy in dilute chloroform solutions and in films spin-coated on quartz substrates. The UV–vis absorption spectra of the polymers in CHCl3 solutions are shown in Fig. 2a. The absorption maxima of the spectra are at 460, 480 and 500 for PT–C1, PT–C2 and PT–C3, respectively. Compared with PT–C1, PT–C2, the red-shift of the absorption maximum for PT–C3 could be attributed to the extension of the conjugated length in PT–C3 with higher molecule weight. In addition, the absorption spectra PT–C2 and PT–C3 solutions show a weak shoulder peak at ca. 610∼620 nm, which indicates that they have good π–π stacking in solution compared to PT–C1. The absorption spectra of the polymer films are shown in Fig. 2b, and the detailed optical data are summarized in Table 2. The absorption maximum of the polymer films all show a large red-shift (>50 nm) compared with that of their solutions, indicating that there is strong intermolecular interaction in the solid state. Furthermore, polymer PT–C3 shows a strong shoulder peak at 600 nm which is similar to P3HT, owing to the ordered alignment of the molecules. The absorption edges (λedge) of the three polymer films are 646 nm for PT–C1, 655 nm for PT–C2 and 650 nm for PT–C3, from which the optical bandgaps (Egopt) of the polymers were calculated to be 1.92, 1.89 and 1.91 eV for PT–C1, PT–C2 and PT–C3 respectively, according to the equation of Egopt = 1240/λedge.
Table 2 Absorption and electrochemical properties of the polymers
|
Polymers
|
UV-vis absorption spectra
|
Cyclic voltammetry
|
| Solutiona |
Filmb |
p-doping |
n-doping |
E
g
EC (eV) |
|
λ
max (nm) |
λ
max (nm) |
λ
onset (nm) |
E
g
opt (eV) c |
φ
ox
/HOMO (V)/(eV) |
φ
red/LUMO (V)/(eV) |
|
Measured in chloroform solution.
Cast from chloroform solution.
Bandgap estimated from the onset wavelength (λedge) of the optical absorption: Egopt = 1240/λedge.
|
|
PT
–C1
|
460 |
520 |
646 |
1.92 |
0.44/−5.15 |
−1.75/−2.96 |
2.19 |
|
PT
–C2
|
480 |
540 |
655 |
1.89 |
0.40/−5.11 |
−1.76/−2.95 |
2.16 |
|
PT
–C3
|
500 |
550 |
650 |
1.91 |
0.39/−5.10 |
−1.73/−2.98 |
2.12 |
Electrochemical properties
Electrochemical cyclic voltammetry was performed to determine the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the conjugated polymers.18Fig. 3 shows the cyclic voltammograms of the PT–C1, PT–C2 and PT–C3 films on Pt disc electrode in 0.1 mol L−1Bu4NPF6 acetonitrile solution. The onset reduction potentials (φred) of PT–C1, PT–C2 and PT–C3 are −1.75, −1.76 and −1.73 V vs. Ag/Ag+, respectively, while the onset oxidation potentials (φox) are 0.44, 0.40 and 0.39 V vs. Ag/Ag+, respectively. From φox and φred of the polymers, HOMO and LUMO energy levels as well as the energy bandgap (EgEC) of the polymers were calculated according to the equations19
| HOMO = −e (φox + 4.71) (eV); |
| LUMO = −e (φred + 4.71) (eV); |
| EgEC = e (φox − φred) (eV) |
Where the units of φox and φred are V vs.Ag/Ag+.
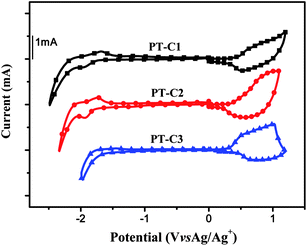 |
| | Fig. 3
Cyclic voltammograms of polymer films on a platinum electrode in 0.1 mol L−1Bu4NPF6 acetonitrile solutions at a scan rate of 100 mV s−1. | |
The results of the electrochemical measurements are also listed in Table 2. It was shown that the LUMO energy levels of the polymers are similar; and the HOMO energy levels of PT–C2 and PT–C3 are a little higher than that of PT–C1, probably due to the longer bithiophene unit in the polymer backbone.20 Obviously, HOMO levels of the polymers significantly decreased from −4.8 eV for P3HT to −5.15, −5.11 and −5.10 eV for PT–C1, PT–C2 and PT–C3, respectively, owing to the electron-withdrawing carboxylate substituent in the side chain of the polymers. It should be noticed that the HOMO level (−5.3 eV) of P3HDTTT15 with a large alkyl side chain is even lower than that of the carboxylate substituted PTs. The lower HOMO of P3HDTTT can be ascribed to less conjugation of the polymer main chain due to the steric hindrance of its big side chain which resulted in poorer absorption of the polymer. For the electron-withdrawing carboxylate substituted PTs, the decrease of the HOMO energy levels is ascribed to the effect of the electron-withdrawing substituent. The same absorption spectra of the polymers with P3HT confirm the high level of conjugation of the polymers with the carboxylate substituent.
Photovoltaic properties
To investigate and compare the photovoltaic properties of the three copolymers, bulk heterojunction PSC devices with a configuration of ITO/PEDOT:PSS/polymers:PC70BM/Al were fabricated. Fig. 4 shows the I–V curves of the PSCs under the illumination of AM1.5, 100 mW cm−2. The open-circuit voltage (Voc), short-circuit current (Jsc), fill factor (FF), and power conversion efficiency (PCE) of the devices (the average values for 5 devices) are summarized in Table 3. And the photovoltaic performance data of the control device based on P3HT:PC70BM with the same device fabrication conditions were also listed on Table 3 for comparison.
Table 3 Photovoltaic performances of the PSCs based on polymer:PC70BM (1:1 w/w) under the illumination of AM1.5, 100 mW cm−2
|
Polymer
|
V
oc (V) |
J
sc (mA cm−2) |
FF |
PCE (%) |
|
The device was fabricated with the same experimental conditions as that used in fabricating the PSCs for the carboxylate substituted PTs.
|
|
PT
–C1
|
0.81 |
5.31 |
0.392 |
1.69 |
|
PT
–C2
|
0.81 |
5.44 |
0.484 |
2.14 |
|
PT
–C3
|
0.77 |
7.22 |
0.584 |
3.25 |
|
P3HT
a
|
0.62 |
7.07 |
0.547 |
2.40 |
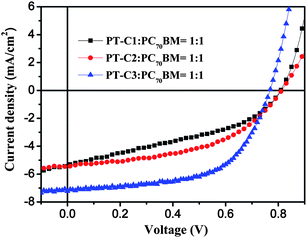 |
| | Fig. 4
J–V curves of the PSCs based on polymer:PC70BM (1:1, w/w), under the illumination of AM 1.5, 100 mW cm−2. | |
The devices based on polymers: PC70BM (1:1, w/w) all exhibit a higher open circuit voltage (0.81V for PT–C1 and PT–C2, 0.77 V for PT–C3) than P3HT (0.62 V), which is benefited from the deep HOMO energy levels of the three polymers.21 The higher Jsc of the device based on PT–C3 than that of the devices based on PT–C1 and PT–C2 agrees with the stronger absorption and higher molecuar weight of PT–C3. The higher molecular weight of the conjugated polymers could lead to higher hole mobility and better charge transportation properties.22 Among the PSCs, the device based on PT–C3:PC70BM (1:1 w/w) displayed the best photovoltaic performance: PCE of 3.25% with a Voc of 0.77 V, an Jsc of 7.22 mA cm−2, and a fill factor (FF) of 0.584.
In order to investigated the effect of morphology on the photovoltaic performance, film morphology of the copolymers:PC70BM (1:1 w/w) was studied by atomic force microscope (AFM). Fig. 5 shows the tapping-mode AFM images. It is clear that only the PT–C3:PC70BM (1:1 w/w) blend film showed a good interpenetrating network which is beneficial to the exciton dissociation and charge carriers transport. Obviously, the suitable morphology of PT–C3:PC70BM film is consistent with the higher photovoltaic performance of the PSCs based on PT–C3.
 |
| | Fig. 5
AFM topography images of the blend films of copolymers: PC70BM (1:1, w/w): (a) PT–C1, (b) PT–C2, (c) PT–C3. | |
In considering the promising photovoltaic properties of PT–C3 among the three copolymers, we performed the optimization of the PSCs based on PT–C3 by changing weight ratios of PT–C3:PC70BM. Fig. 6 shows the current–voltage characteristics of the PSC devices at different device fabrication conditions and Table 4 lists the photovoltaic properties of the devices. The best photovoltaic performance with the maximum PCE value of 3.87% (Voc = 0.78 V, Jsc = 9.68 mA cm−2, FF = 51.2%) was obtained in the PSC device with a weight ratio of PT–C3:PC70BM = 1:1.5. In comparison with the PSC based on PT–C3:PC70BM = 1:1.5, lower PC70BM weight ratio at PT–C3:PC70BM = 1:1 led to lower Jsc (7.22 mA cm−2), due probably to inefficient charge separation and lower electron transporting efficiency.23 While higher PC70BM weight ratio at PT–C3:PC70BM = 1:2 w/w also resulted in lower Jsc (6.43 mA cm−2). The lower Jsc at higher PC70BM content could be ascribed to the reduction of the absorption of PT–C3 and an unbalanced charge transporting properties between holes and electrons due to the higher PC70BM content.24
Table 4 Photovoltaic properties of the PSCs based on PT–C3:PC70BM with different weight ratios, under the illumination of AM1.5, 100 mW cm−2
| Active layer |
V
oc (V) |
J
sc (mA cm−2) |
FF |
PCE (%) |
|
PT
–C3:PC70BM = 1:1 |
0.77 |
7.22 |
0.584 |
3.25 |
|
PT
–C3:PC70BM = 1:1.5 |
0.78 |
9.68 |
0.512 |
3.87 |
|
PT
–C3:PC70BM = 1:2 |
0.82 |
6.43 |
0.529 |
2.79 |
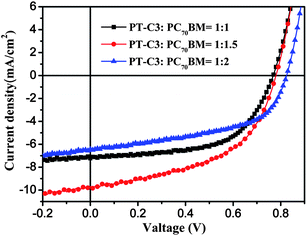 |
| | Fig. 6
J–V curves of the PSCs based on PT–C3:PC70BM with different weight ratios under the illumination of AM 1.5, 100 mW cm−2. | |
As shown in Fig. 7, the external quantum efficiency (EQE) curves of the PSCs based on PT–C3:PC70BM with different weight ratios all covers a broad wavelength range from 300 to 700 nm. The device with weight ratio of 1:1.5 exhibited the higher EQE in the absorption range in comparison with the devices with the weight ratios of 1:1 and 1:2. The maximum EQE value of the device based on PT–C3:PC70BM (1:1.5, w/w) reached 57.5% at ca. 470 nm. The short-circuit current density obtained from integrating the EQE curves of PT–C3 blends is 9.2 mA cm−2, which reasonably agree to the measured Jsc of 9.68 mA cm−2 under the illumination of AM1.5, 100 mW cm−2.
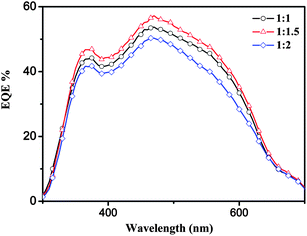 |
| | Fig. 7 EQE of the PSCs based on PT–C3: PC70BM with different weight ratios. | |
Conclusion
In summary, we synthesized three polythiophene derivatives PT–C1, PT–C2 and PT–C3 with carboxylate substituent by Stille-coupling reaction. The polymers have a strong absorption at 350–650 nm, and exhibit good solubility and thermal stability. By attaching the carboxylate substituent in the side chain, the HOMO energy level of the polymers is successfully decreased from ca. −4.8 eV for P3HT to −5.15, −5.11 and −5.10 eV for PT–C1, PT–C2 and PT–C3, respectively. The devices based on the blend of the three polymers and PC70BM all exhibit relatively high open circuit voltage of ca. 0.8 V. PCE of the PSCs based on PT–C3: PC70BM = 1:1.5(w/w) reached 3.87% under the illumination of AM1.5, 100 mW cm−2, with Voc = 0.78 V, a short circuit current of 9.68 mA cm−2, and a fill factor of 51.2%. These results indicate that attaching the carboxylate substituent is a simple and effective way to modulate energy levels of polythiophene derivatives and PT–C3 is a promising material for the polymer solar cells.
Experimental section
Materials
All chemicals and solvents were reagent grades and purchased from Aldrich, Alfa Aesar and TCI Chemical Co. Toluene, tetrahydrofuran, and diethyl ether were distilled to keep anhydrous before use. All of the other chemicals were used as received.
Measurements and characterization
All new compounds were characterized by 1H NMR spectroscopy performed on a Bruker DMX-400 spectrometer. For the 1H-NMR measurements, CDCl3 was used as the solvent. Chemical shifts in the NMR spectra were reported in ppm relative to the singlet at 7.26 ppm for CDCl3. The molecular weight of the polymers was measured by gel permeation chromatography (GPC), and polystyrene was used as a standard. Thermogravimetric analysis (TGA) was performed on a Perkin–Elmer TGA-7. Mass spectra were obtained with a Shimadzu QPT-C2010 spectrometer. UV-Vis absorption spectra were obtained on a Hitachi U-3010 spectrometer. Cyclic voltammetry (CV) was performed on a Zahner IM6e Electrochemical Workstation with a three-electrode system in a solution of 0.1 M Bu4NPF6 in acetonitrile at a scan rate of 100 mV s−1. The polymer films were coated on a Pt plate electrode by dipping the electrode into the corresponding solutions and then drying. A Pt wire was used as the counter electrode, and Ag/Ag+ was used as the reference electrode.
Device fabrication and characterization of polymer solar cells
Polymer solar cells (PSCs) were fabricated with ITO glass as a positive electrode, Al as a negative electrode and the blend film of the polymer/PC70BM between them as a photosensitive layer. The ITO glass was precleaned and modified by a thin layer of PEDOT:PSS which was spin-cast from a PEDOT:PSS aqueous solution (Clevious P VP AI 4083 H. C. Stark, Germany) on the ITO substrate, and the thickness of the PEDOT:PSS layer is about 60 nm. The photosensitive layer was prepared by spin-coating a blend solution of polymers and PC70BM in o-dichlorobenzene on the ITO/PEDOT:PSS electrode. Then the Al cathode was deposited on the polymer layer by vacuum evaporation under 5 × 10−5 Pa. The thickness of the photosensitive layer is ca.120∼150 nm, measured on an Ambios Tech. XP-2 profilometer. The effective area of one cell is 4 mm2. The current–voltage (I–V) measurement of the devices was conducted on a computer-controlled Keithley 236 Source Measure Unit. A xenon lamp with AM1.5 filter was used as the white light source, and the optical power at the sample was around 100 mW cm−2.
Synthesis
The compounds 2, 5-bis(trimethylstannyl)thiophene24 (4), 5,5′-bis(trimethyl-stannyl)-2,2′-bithiophene25 (5) and thiophene-3-carboxylic acid26 were synthesized according to the procedures in the literature. The synthetic routes for the monomer and polymers are shown in Scheme 2. The detailed synthetic processes are as follows.
2,5-Dibromothiophene-3-carboxylic acid
27 (1).
Thiophene-3-carboxylic acid (1) (1.28 g, 0.01 mol) was added to glacial acetic acid (25 mL). Then Br2 (2.56 mL, 0.05 mol) was added to the flask dropwise at room temperature. The mixture was warmed and stirred at 60 °C for 15 h. After the reaction was completed, the mixture was poured into 200 mL of deionized water, and some Na2SO3 was added to decolorize. After dehydration and evaporation of the solvent, the crude product was purified by recrystallization from EtOH-H2O to give product 2 as white needles (2.49 g, yield 87%); mp 178 °C. 1H NMR (300 MHz, DMSO-d6, δ (ppm)): 13.31 (s, 1H), 7.40 (s, 1H). 13C NMR (100 MHz, CDCl3, δ (ppm)): 164.8, 131.6, 130.5, 120.9, 110.9. ESI: m/z = 284.
2-Ethylhexyl-2,5-dibromothiophene-3-carboxylate
28 (2).
To the mixture of 2,5-dibromothiophene-3-carboxylic acid (2.86 g, 10.0 mmol), DCC (1.2 g, 12 mmol), DMAP (0.32 g, 3.5 mmol) in a 100 mL round-bottom flask with 50 mL CH2Cl2 were added 2-ethylhexan-1-ol (6.5 g, 50 mmol). The mixture was stirred for 40 h under N2 atmosphere, was poured into 30 mL water, and then extracted with CH2Cl2. The organic phase was dried with sodium sulfate and the solvent was removed. The product was purified with column chromatography on silica gel using hexane/CH2Cl2 (4:1), yielding the pure compound as a colorless oil 3.66 g (92%). EI: m/z = 398. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.31(s, 1H), 4.20–4.18 (d, 2H), 1.73–1.71 (m, 1H), 1.35–1.23 (m, 8H), 0.89–0.86 (m, 6H). 13C NMR (100 MHz, CDCl3, δ (ppm)): 160.94, 132.29, 118.91, 111.43, 77.60, 77.28, 76.97, 67.63, 38.94, 30.64, 29.10, 24.06, 23.12, 14.21, 11.21. Anal. Calcd for C13H18Br2O2S: C, 39.22; H, 4.56; Br, 40.14; O, 8.04; S, 8.05. Found: C, 39.01; H, 4.78.
2-Hexyldecyl-2,5-dibromothiophene-3-carboxylate (3).
The compound 3 was synthesized according to the similar procedure with compound 2. 2,5-Dibromothiophene-3-carboxylic acid (2.86 g, 10.0 mmol), DCC (1.2 g, 12 mmol), DMAP (0.32 g, 3.5 mmol) and 3-hexyldecan-1-ol (12.1 g, 50 mmol). After workup, the pure product was obtained as a colorless oil, 4.8 g (94%). EI: m/z = 508.
1H NMR (CDCl3, 400 MHz), δ (ppm): 7.33 (s, 1H), 4.19–4.18 (d, 2H), 1.73–1.71 (m, 1H), 1.37–1.25 (m, 24H), 0.89–0.86 (m, 6H). 13C NMR (100 MHz, CDCl3, δ (ppm)): 181.06, 132.31, 131.94, 118.92, 111.44, 77.51, 77.40, 77.19, 76.87, 68.13, 37.47, 32.05, 31.96, 31.49, 30.07, 29.74, 29.70, 29.61, 26.86, 22.83, 22.80, 14.25. Anal. Calcd for C21H34Br2O2S: C, 49.42; H, 6.71; Br, 31.31; O, 6.27; S, 6.28.Found: C, 49.21; H, 6.92.
Synthesis of polymer PT–C1.
Compound 2 (199 mg, 0.5 mmol), compound 4 (206 mg, 0.5 mmol), and dry toluene (12 mL) were added to a 50 mL double-neck round-bottom flask. The reaction container was purged with argon for 20 min to remove O2, and then Pd (PPh3)4 (15 mg) added. After another flushing with argon for 20 min, the reactant was heated to reflux for 12 h. The reactant was cooled down to room temperature and poured into MeOH (200 mL), then filtered through a Soxhlet thimble, which was then subjected to Soxhlet extraction with methanol, hexane, and chloroform. Polymer was recovered from the chloroform fraction by rotary evaporation as solid. The polymer was purified by chromatography on silica gel with chloroform as the eluent, and the polymer solution was concentrated and was poured into MeOH. After this, the precipitates were collected and dried under vacuum overnight. Yield: 108 mg (68%). GPC: Mw = 6.9 K; Mn = 4.3 K; Mw/Mn = 1.62. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.52–7.33 (m, 1H), 7.03–6.87 (m, 4H), 4.20 (s, 2H), 1.70–0.88 (m, 31H).
Synthesis of polymer PT–C2.
PT
–C2 was synthesized following the same procedure as that in the synthesis of PT–C1 except that compound 5 was used instead of compound 4. Yield: 142 mg (71%). GPC: Mw = 5.6 K; Mn = 3.2 K; Mw/Mn = 1.75. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.60–7.48(m, 1H), 7.08–6.73 (m, 4H), 4.21 (s, 2H), 1.70–0.92 (m, 15H).
Synthesis of polymer PT–C3.
PT
–C3 was synthesized following the same procedure as that in the synthesis of PT–C2 except that compound 3 was used instead of compound 2. Yield: 210 mg (82%). GPC: Mw = 113.4 K; Mn = 69.4 K; Mw/Mn = 1.63. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.57–7.48 (m, 2H), 7.20–7.05 (m, 1H), 4.21 (s, 2H), 1.70–0.91 (m, 15H).
Acknowledgements
This work was supported by NSFC (Nos. 20874106, 20821120293, 50933003 and 21021091), the Ministry of Science and Technology of China and Chinese Academy of Sciences.
References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
-
(a) H.-Y. Chen, J. H. Hou, S. Q. Zhang, Y. Y. Liang, G. W. Yang, Y. Yang, L. P. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS;
(b) Y. Y. Liang, Z. Xu, J. B. Xia, S.T. –Tsai, Y. Wu, G. Li, C. Ray and L. P. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS.
-
(a) J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS;
(b) G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS;
(c) Y.-J. Chen, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef;
(d) Y. F. Li and Y. P. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS.
-
(a) Y. P. Zou, A. Najari, P. Berrouard, S. Beaupré, B. R. Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS;
(b) T. Chu, J. Lu, S. Beaupr, Y. Zhang, J. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS.
-
(a) G. Li, V. Shrotriva, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS;
(b) W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- J. H. Hou, Z. A. Tan, Y. Yan, Y. J. He, C. H. Yang and Y. F. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS.
-
(a) Y. J. He, H.-Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS;
(b) Y. J. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- G. J. Zhao, Y. J. He and Y. F. Li, Adv. Mater., 2010, 22, 4355–4358 CrossRef CAS.
- I. Osaka, R. Zhang, G. Sauve, D. M. Smilgies, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2009, 131, 2521 CrossRef CAS.
-
(a) D. H. Kim, B.-L. Lee, H. Moon, H. M. Kang, E. J. Jeong, J.-L. Park, K.-M. Han, S. Lee, W. B. Yoo, B. W. Koo, Y. Kim, W. H. Lee, K. Cho, H. A. Becerril and Z. Bao, J. Am. Chem. Soc., 2009, 131, 6124 CrossRef CAS;
(b) M. J. Zhang, H. J. Fan, X. Guo, Y. J. He, Z. G. Zhang, J. Min, J. Zhang, G. J. Zhao, X. W. Zhan and Y. F. Li, Macromolecules, 2010, 43, 5706–5712 CrossRef CAS.
-
(a) Y. Zhu, R. D. Champion and S. A. Jenekhe, Macromolecules, 2006, 39, 8712–8719 CrossRef CAS;
(b) P. T. Wu, F. S. Kim, R. D. Champion and S. A. Jenekhe, Macromolecules, 2008, 41, 7021–7028 CrossRef CAS.
-
(a) X. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206 CrossRef CAS;
(b) H. Xin, X. Guo, F. S. Kim, G. Ren, M. D. Watson and S. A. Jenekhe, J. Mater. Chem., 2009, 19, 5303 RSC.
- M. C. Yuan, M. Y. Chiu, S. P. Liu, C. M. Chen and K. H. Wei, Macromolecules, 2010, 43, 6936–6938 CrossRef CAS.
- J. H. Hou, H. Y. Chen, S. Q. Zhang, L. J. Huo, S. Sista and Y. Yang, Macromolecules, 2009, 42, 9217–9219 CrossRef CAS.
- L. J. Huo, T. L. Chen, Y. Zhou, J. H. Hou, H. Y. Chen, Y. Yang and Y. F. Li, Macromolecules, 2009, 42, 4377–4380 CrossRef CAS.
- X. L. Hu, M. M. Shi, J. Chen, L. J. Zuo, L. Fu, Y. J. Liu and H. Z. Chen, Macromol. Rapid Commun., 2011, 32, 506–511 CAS.
- Y. F. Li, Y. Cao, J. Gao, D. L. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243–248 CrossRef CAS.
- Q. J. Sun, H. Q. Wang, C. H. Yang and Y. F. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- K. C. Li, J. H. Huang, Y. C. Hsu, P. J. Huang, C. W. Chu, J. T. Lin, K. C. Ho, K. H. Wei and H. C. Lin, Macromolecules, 2009, 42, 3681–3693 CrossRef CAS.
- M. H. Tong, S. Cho, J. T. Rogers, K. Schmidt, B. Hsu, D. Moses, R. C. Coffin, E. J. Kramer, G. C. Bazan and A. J. Heeger, Adv. Funct. Mater., 2010, 20, 3959–3965 CrossRef CAS.
-
(a) P. A. C. Quist, T. J. Savenije, M. M. Koetse, S. C. Veenstra, J. M. Kroon and L. D. A. Siebbeles, Adv. Funct. Mater., 2005, 15, 469–474 CrossRef CAS;
(b) N. S. Baek, S. K. Hau, H. L. Yip, O. Acton, K. S. Chen and A. K. Y. Jen, Chem. Mater., 2008, 20, 5734–5736 CrossRef CAS.
- J. H. Huang, Z. Y. Ho, D. Kekuda, Y. Chang, C. W. Chu and K. C. Ho, Nanotechnology, 2009, 20, 025202 CrossRef.
-
(a) D. E. Seitz, S. H. Lee, R. N. Hanson and J. C. Bottaro, Synth. Commun., 1983, 13, 121 CrossRef CAS;
(b) C. V. Pham, R. S. Macomber, H. B. Mark and H. Zimmer, J. Org. Chem., 1984, 49, 5250–5253 CrossRef.
-
(a) H. Goto and K. Akagi, Angew. Chem., Int. Ed., 2005, 44, 4322 CrossRef CAS;
(b) H. H. Fong, V. A. Pozdin, A. Amassian, G. G. Malliaras, D. M. Smilgies, M. Q. He, S. Gasper, F. X. Zhang and M. Sorensen, J. Am. Chem. Soc., 2008, 130, 13202–13203 CrossRef CAS.
- E. Campaigne and W. M. Lesuer, J. Am. Chem. Soc., 1948, 70, 1555 CrossRef CAS.
- E. Campaigne and R. C. Bourgeois, J. Am. Chem. Soc., 1954, 76, 2445 CrossRef CAS.
- Y. Y. Liang, S. Q. Xiao, D. Q. Feng and L. P. Yu, J. Phys. Chem. C, 2008, 112, 7866–7871 CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.