Investigation into fiber formation in N-alkyl urea peptoid oligomers and the synthesis of a water-soluble PEG/N-alkyl urea peptoid oligomer conjugate†
Xiaoping
Chen
,
Keyang
Ding
and
Neil
Ayres
*
Department of Chemistry, The University of Cincinnati, PO Box 210172, Cincinnati, Ohio 45221, USA. E-mail: Neil.Ayres@UC.edu; Fax: (+513) 556-9239; Tel: (+513) 556-9280
First published on 30th August 2011
Abstract
The synthesis and self assembly of an N-alkyl urea peptoid oligomer/polymer conjugate that self-assembles into ribbon-like structures is reported. The aggregation of the N-alkyl urea peptoid oligomers has been investigated using NMR spectroscopy, with long and highly uniform fibers with micron dimensions observed in scanning electron microscopy images from both organo-soluble and water-soluble versions. The results of these studies indicate that hydrogen bonding likely is the major driving force for the self assembly. The oligomer was conjugated to poly(ethylene glycol) (PEG) producing a water soluble polymer/N-alkyl urea peptoid conjugate where the self-assembly of the N-alkyl urea peptoid was retained. These polymer conjugates represent a new approach to polymer biomimetic conjugates.
Introduction
Polymer bioconjugates are macromolecules that contain synthetic polymers and monodisperse biological segments.1–4 The polymer segments enhance the physical or mechanical properties of the bioconjugates,5,6 while the biological segments provide self-assembly or impart biological activities.7–10 Various morphologies have been obtained from self-assembled bioconjugates, such as fibers,11–13 3-dimensional hydrogels,14–16 nanosponges,17–19 and microgels/nanogels.20,21 Self-assembling bioconjugates have the potential to benefit many diverse applications, including therapeutics,22 gene delivery,23 and biosensors.24 Controlled/“living” or “reversible deactivation radical”25 polymerization techniques, in addition to ring opening metathesis polymerization (ROMP), have been used to prepare synthetic polymer segments in bioconjugates.26–31 These polymers can possess pre-determined molecular weights and narrow molecular weight distributions, and include a variety of potential functional groups, many of which are stimuli-responsive, thereby imparting additional properties into the bioconjugates. Synthesis of bioconjugates using controlled/“living” polymerization is achieved through two different approaches. The first method is known as “grafting to”, and involves the reaction of functional groups on the peptide or protein with appropriate functional groups present in the synthetic polymer, either as end groups or side chains. This can be considered as post-modification of polymers with the naturally occurring molecule. In the second method, called “grafting from”, a small molecule (i.e. an NMP initiator, ATRP initiator, or RAFT agent) is attached to the peptide or protein followed by the polymer propagation reaction. For example, Ting et al.32 reported the synthesis of lectin recognizable biomaterials via NMP using the “grafting to” method. Alternatively, protein functionalized nanogels have been prepared using ATRP in inverse miniemulsion process by the “grafting from” method.33 The synthesis of bioconjugates using RAFT polymerization was highlighted by a recent review by Davis et al.34As part of our continuing goal to prepare synthetic analogues of bioconjugated polymers, we have become interested in peptidomimetics. Peptidomimetics are non-natural oligomers that are inspired by naturally occurring oligomers, and have the ability to self-assemble or fold into patterns such as helices, turns, and strands. Oligomers containing urea-functional backbones are one example these molecules, and were highlighted in a recent perspective by Fischer and Guichard.35 These N,N′-linked urea oligomers demonstrate a propensity to fold into helical structures in solution due to their hydrogen-bonding properties,36–40 and have been well characterized using both NMR spectroscopy36,40,41 and X-ray crystallography.42 The oligoureas are attractive for biomedical applications due to the resistance of the urea group to enzymatic degradation.43 For example, oligoureas have been designed that possess antimicrobial properties.44,45 Recently, we reported the synthesis of an N-alkyl-N,N′-linked urea oligomer, and its coupling to well-controlled polymers prepared using reversible addition fragmentation chain transfer (RAFT) polymerization.46 We referred to our oligomer as an N-alkyl urea peptoid based on the work of Zuckermann and others with N-substituted glycine peptidomimetics, which are commonly called “peptoids”.47–49 Subsequently, we prepared a low grafting density molecular brush from a poly(N-alkyl urea peptoid) core using a step-growth polymerization, where we observed that the poly(N-alkyl urea peptoid) formed ill-defined fibers in dichloromethane.50 We hypothesized that the self-assembly of the poly(N-alkyl urea peptoid) was due to interactions of the N-alkyl urea peptoid monomers along the polymer backbone. The monomer contained a modified version of N-alkyl urea peptoid oligomer 1 (Fig. 1), containing six urea groups.
 | ||
| Fig. 1 Structure of oligomer 1. | ||
We incorporated two 2,4-dimethoxybenzyl (DMB) groups into the oligomer, often used in peptide synthesis to inhibit hydrogen bonding and intermolecular folding or self-assembly. We used this moiety in the synthesis of oligomer 1 for the same purpose, i.e. to prevent self-assembly; therefore, we were surprised to see the aforementioned fibers.
Herein, we report our investigations into the self-assembly of the N-alkyl urea peptoid oligomer, and furthermore, show the self-assembly is retained when the oligomer is conjugated to poly(ethylene glycol) (PEG), which we have called the PEGylated conjugate. The PEGylated conjugate represents a completely synthetic analog to PEG-peptide “chimera” polymer bioconjugates.
Results and discussion
The poly(N-alkyl urea peptoid) backbone was observed to self-assemble in organic solution into fiber-like structures. Therefore, we hypothesized that we should also observe the formation of aggregates from oligomer 1, as it seemed likely that the N-alkyl urea peptoid segments caused the poly(N-alkyl urea peptoid) self-assembly, rather than the diisocyantate used as the comonomer in the step polymerization.50 Furthermore, examination of self-assembly of the monomeric N-alkyl urea peptoid 1 should be easier to perform than for the polymer; therefore we directed our attention to the oligomer. Indeed, we observed that fibers formed in a 10 mg ml−1 solution of 1 in CH2Cl2 after the solution was allowed to rest at room temperature for 72 h. Scanning electron microscopy (SEM) revealed the aggregates to be long, linear fibers of highly disperse width and length (Fig. 2). | ||
| Fig. 2 SEM images of fibers resulting from the self-assembly of 1 from CH2Cl2. | ||
We initially used dichloromethane as the aggregates arising from the poly(N-alkyl urea peptoid) were observed to form in this solvent. We next examined N,N-dimethylformamide (DMF) as a solvent, reasoning that the increased polarity of DMF could lead to different results than those obtained in dichloromethane. SEM images of aggregates formed in a solution of 1 in DMF at a concentration of 1 mg ml−1 using the same conditions showed fibers possessing uniform diameters and much longer lengths (Fig. 3). A lower concentration compared to the dichloromethane (DCM) solution was used due to the oligomers having lower solubility in DMF compared to DCM. Examination of the SEM image showed the diameters of the fibers to be around 9.4 μm.
 | ||
| Fig. 3 SEM images of fibers resulting from the self-assembly of 1 in DMF. (a) Fibers of 1 self assembled in DMF. (b) Magnification of fiber marked with arrow in Fig. 3a. | ||
We used 2-dimensional Nuclear Overhauser Effect (NOE) correlation NMR experiments (NOESY) in CD2Cl2 at a concentration below that where fiber formation is observed (Fig. 4) to probe the molecular interactions. The lower concentration was used to ensure that the oligomers remained in solution during the experiment. NOE correlations have been used in determining the helical structure of urea oligomers,36 determining β-sheet structures of short synthetic peptide oligomers,51,52 and for observation of parallel sheet self-assembly of γ-peptides.53 Our N-alkyl urea peptoid oligomers are similar to the γ-2-peptides, where our molecules posses a nitrogen atom at the site of the α-C in the peptide.54,55
 | ||
| Fig. 4 (a) Key part of the NOESY spectrum of N-alkyl urea peptoid 1 at a concentration of 10 mg ml−1 in CD2Cl2. (b) Structure of compound 1 with hydrogen atoms labeled corresponding to Figure 4a and 4d. (c) Through space couplings indicated by the NOESY spectrum on two molecules of 1. The through space couplings are shown by dashed double-headed arrows and DMB = 2,4-Dimethoxybenzene. (d) Key part of the NOESY spectrum of N-alkyl urea peptoid 1 at the concentration of 4 mg ml−1 in CD2Cl2. | ||
Examination of the NOE correlation spectrum (Fig. 4a) revealed that the first and fourth N-methyl groups (i.e. those furthest from the center of the oligomer and the DMB groups) couple to the ortho proton of a nitrosulfobenzyl group (Ns) and a proton in a DMB group. In contrast, the “interior” N-methyl groups (i.e. those closest to the center of the oligomer and the DMB groups) coupled with the DMB moiety and a urea proton. The through space couplings are shown in Fig. 4c, however, we are not claiming that the oligomers are arranged in an “as drawn”, extended “all-trans” conformation. The larger, circular coupling peak in the spectrum arises from coupling between an N–H proton and the N–CH3 group. The N–H proton is unclear at the magnification of the proton spectra on the top x-axis in Fig. 4a. The –OCH3 groups are equivalent in the NOESY spectrum, however, we have not indicated through space coupling to these groups from the N-CH3 group on an adjacent molecule in Fig. 4c for clarity purposes. The N–CH3 groups are separated from the aromatic groups by a distance of at least 7 bonds, a distance where through-space NOE coupling should be undetectable if these molecules are somewhat extended (i.e., not folded). Therefore, the observed NOESY correlations would likely be intermolecular in nature. However, these through space couplings would be highly likely to be intra-molecular in nature if the N-alkyl urea peptoid was folding upon itself – and there should be enough flexibility within the molecule from the ethylene bridges between urea groups to permit this. In order to test this, we repeated the NOESY experiment with a 2.5-fold dilution in concentration from the original solution, giving a concentration of 4 mg ml−1 (Fig. 4c). We also extended the NOESY experiment time from 12 h to 24 h to account for the lower concentration. The total disappearance of two cross peaks from the methyl group at 2.88 ppm, and dramatic reduction in peak intensity from all other interactions is observed when comparing Fig. 4d with Fig. 4a. (The peak at ∼ 5.3 ppm vs. 2.94 ppm in Fig. 4d is ascribed to noise from the experiment at high magnification of the spectrum). This implies that the NOESY results from Fig. 4a involve coupling between molecules, rather than arising from discrete, folded, molecules. If we were observing single molecules folding upon themselves the reduction/disappearance of peaks in the NOESY spectrum should not be so pronounced.
Initially, we assumed that a combination of hydrogen bonding and π–π interactions between the aromatic moieties in 1 were responsible for the observed self-assembly. These interactions are known to be important in non-covalent self-assembly processes in both biology and chemistry. A classical example of where such interactions play crucial roles is low molecular weight organo-gelators, or “organogels”, where several systems incorporate the urea functional group.56–59 (It should be noted that the aggregates we observed precipitated from organic solution as fibers rather than afforded any gel-like behavior.) However, the spectra in Fig. 4 do not support the presence of aromatic interactions between oligomers of 1 under the conditions of the NOESY experiment. If π–π stacking interactions were present between the DMB groups and the Ns protecting groups, cross peaks should be observed between these groups in the NOESY spectra. However, in the aromatic region, only two strong cross peaks are present: one is between the intra-protons (∼8.04ppm, ∼7.72 and ∼7.70ppm) of the Ns protecting group and the other is between the intra-protons (∼6.96ppm, ∼6.44ppm and ∼6.47ppm) of the DMB group, respectively. This means that the Ns groups and DMB groups are a distance further apart than can be probed by the NOESY experiment. We have made the reasonable assumption that any π–π stacking distances should be of the same magnitude as the NOE correlations. Conversely, if interactions were present both from π–π stacking between DMB groups present on different molecules, and from π–π stacking between Ns groups from different molecules respectively (i.e. similar to two N-alkyl urea peptoids pressed together), the intra- and the inter-molecular NOE peaks in the aromatic region would be degenerate and overlap together. However, these interactions would require the molecules to be parallel and aligned without substantial displacement from each other. As we described above, this is not observed in the NOESY spectra we obtained. Additionally, we did not observe any change in the peak shape of the UV-Vis spectrum of 1 at different concentrations (spectra available in the ESI†), starting from a concentration close to where fibers precipitated from a 1 mg ml−1 solution of oligomer 1 and decreasing with serial dilutions. In contrast, Song and co-workers60 observed that the peak shape in UV-Vis spectra of an amphiphile that assembled into nanofibers changed as a result of π–π-interactions driving fiber assembly. Collectively, these results suggest minimal contributions from the aromatic groups in the aggregation of oligomer 1 at the concentrations of the NMR experiments. We have not eliminated the possibility that the aromatic groups aid in bringing individual aggregates together in solution at higher concentrations to form the precipitated fibers, as these NMR experiments have been performed at concentrations below that where we see fiber formation, however, fibers formed only with oligomer 1, not shorter oligomers—all of which possessed the same four aromatic groups as 1.
We are not able to use Circular Dichroism (CD) spectroscopy to investigate potential structure formation or folding due to the fact that these oligomers are not chiral, and therefore would not afford a signal in a CD spectrum. Indeed, due to the divergent synthesis strategy used for these molecules there is a mirror plane at the center of the molecule—hence the use of chiral side groups would still not permit use of CD spectroscopy.
We next synthesized the water-soluble N-alkyl urea peptoid 3 from N-alkyl urea peptoid 1 (Fig. 5) and examined the self-assembly of oligomer 3, which does not possess any aromatic groups.
 | ||
| Fig. 5 Synthesis of the water-soluble N-alkyl urea peptoid oligomer 3. | ||
Oligomer 3 is prepared from oligomer 1 by removal of the 2-nitrobenzenesulfonyl and 2,4-dimethoxybenzyl aromatic groups. The structure of oligomer 3 was confirmed by 1H NMR spectroscopy by observation of the complete disappearance of aromatic proton signals, and using mass spectrometry where a molecular ion peak was observed at m/z = 633.43 (calculated m/z = 633.45 M + 1). Oligomer 3 is a water soluble molecule, and long uniform fibers were observed in SEM images that precipitated from a 10 mg ml−1 aqueous solution of 3 after standing at room temperature for 72 h (Fig. 6).
 | ||
| Fig. 6 SEM images of fibers resulting from the self-assembly of 3. (a) Fibers of 3 self assembled in water. (b) Magnification of fiber marked with arrow in Fig. 6a. | ||
This result gives further weight to the hypothesis that π–π stacking does not appear to be crucial for fiber formation and precipitation of N-alkyl urea peptoids. However, while the similar observations of fibers of 3 forming in water and fibers of 1 forming in organic solution suggest similar assembly mechanisms, the fact 3 possesses secondary urea and primary amine functionality that 1 does not could introduce new driving forces for self-assembly. We ensured that the amines were uncharged following the acidic deprotection reaction, as protonation of the amine groups below their pKa should prevent aggregation due to electrostatic repulsions. Indeed, addition of a few drops of TFA to fibers formed from oligomer 3 in aqueous solution resulted in complete dissolution of the fibers. We suggest that this is a result of the TFA synergistically disrupting hydrogen bonding between oligomers and inducing electrostatic repulsions between charged amine groups. We are unable to perform meaningful NOESY experiments with oligomer 3 as cross-coupling peaks would arise from adjacent groups, obscuring any structural information that could be found.
Having discounted π–π stacking as a major driving force for fiber formation in either the organo-soluble or water-soluble oligomers, and considering the urea functionality in these molecules, the self-assembly interactions are likely to include hydrogen bonding. To examine this further we performed a “D2O shake” test on oligomer 1 in CD2Cl2 to look for H/D exchange (Fig. 7). In addition to hydrogen bonding however, the observation that oligomer 3 self-assembles in water implies that other driving forces must be present beyond only hydrogen bonding, i.e. hydrophobic interactions.
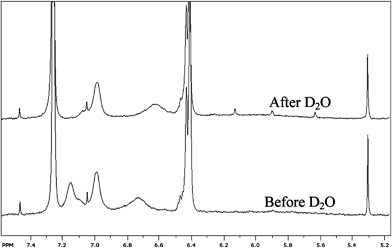 | ||
| Fig. 7 1H NMR spectra of Oligomer 1 in CD2Cl2 before and after addition of D2O. The lower spectrum corresponds to the sample before addition of D2O, the upper spectrum corresponds to the sample after addition of D2O. The initial concentration of 1 in CD2Cl2 was 10 mg ml−1. | ||
We used sufficient D2O to yield a final D2O/CD2Cl2 ratio (v/v) of 1/5. Inspection of Fig. 7 shows the disappearance of the N–H proton peak at ∼7.15 ppm after the D2O shake, meaning that the hydrogen has exchanged with a deuterium. Conversely, the N–H proton peak at 7.0 ppm is unchanged; likewise the N–H peak at ∼6.7 ppm is still present, albeit shifted to ∼ 6.6 ppm. These results demonstrate that while the N–H hydrogen with a peak at 7.15 ppm is accessible to the D2O, the N–H protons giving rise to peaks at 7.0 and ∼6.7 are prevented from exchanging with deuterium atoms during the time-frame of the D2O shake. It seems unlikely that these relatively small molecules would be solvent-inaccessible as might be expected with globular proteins, but rather that these hydrogen atoms are engaged in hydrogen bonding. This supports our argument that the N-alkyl urea peptoids are aggregating using hydrogen bonding. Interestingly, the N–H proton that we observe to exchange with D2O is not from the sulfonamide Ns group, as might be expected if these results correlated to an assumed difference in behavior of the two protons (i.e. urea vs. sulfonamide). In order to provide corroborating evidence for hydrogen bonding interactions we also performed a variable temperature NMR study on oligomer 1. We performed these experiments in CDCl3 as CD2Cl2 cannot be used over the whole temperature range of the study. This lead to some of the peaks slightly changing position from where we described the spectra in CD2Cl2. The rationale for this experiment is that the resonance of most signals is not greatly affected by temperature, although OH, NH, and SH protons can resonate at a higher field at higher temperatures because the degree of hydrogen bonding is reduced.61 The results of the temperature study (shown in ESI,† Fig. S4) show the N–H resonances at 7.4 and ∼6.9 ppm at 275 K shift to higher field with increasing temperature. The two peaks shifting to higher fields imply the presence of hydrogen bonding in these samples. We mirrored the variable temperature study by performing a variable concentration 1H NMR spectra study using serial dilutions (data not shown); however, we observed no significant changes of peak shift or peak shape over the concentration range studied. This result is not too surprising considering that fibers formed at concentrations greater than those used in the NMR experiment, and that pulsed gradient spin echo NMR data (not shown) did not suggest the presence of large-sized aggregates present in solution at our NMR experiment conditions.
PEGylation is the process of attaching molecules to polyethylene glycol through covalent bonds.62 Börner's group,1,63,64 among others,4 reported the successful synthesis of bioconjugates through PEGylation of peptide sequences. These bioconjugates can self-assemble after being triggered by a stimulus such as pH change or deprotecting of phosphate side groups by enzyme digestion. In our work, PEGylation has been performed to extend the scope of the self-assembling N-alkyl urea peptoid oligomers into applications including producing novel types of biocompatible conjugates, and biorelated and biomimetic materials. We have coupled PEG to N-alkyl urea peptoid oligomer 1 using the Fukuyama reaction65 with a commercial PEG methyl ether tosylate. The PEG methyl ether tosylate was of relatively low molecular weight, possessing a number average molecular weight (Mn) of 1200 g mol−1, weight-average molecular weight (Mw) of 1240 g mol−1, and polydispersity (PDI) of 1.03 as measured by GPC calibrated against poly(methyl methacrylate) (PMMA) standards. Every N-alkyl urea peptoid oligomer contains two sulfonamide groups, thus the resulting PEG conjugate possesses two PEG segments at both ends with the N-alkyl urea peptoid oligomer in the center. Therefore the PEGylated conjugate, 4, can be thought of as an ABA triblock copolymer (Fig. 8).
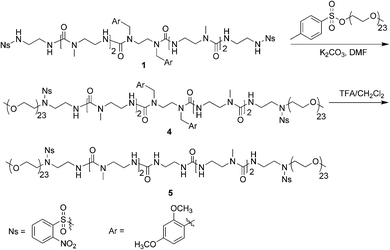 | ||
| Fig. 8 Synthetic scheme of PEG conjugates 4 and 5. | ||
The successful formation of the PEG-b-N-alkyl urea peptoid-b-PEG conjugate 4 was confirmed by 1H NMR spectroscopy and gel permeation chromatography (GPC). The 1H NMR spectrum of conjugate 4 showed sharp peaks at 3.65 ppm belonging to the OCH2 groups of the PEG segments, and at 3.79 ppm corresponding to the OCH3 protons of the DMB groups. The linear conjugate 4 possessed an Mn of 3700 g mol−1, an Mw of 3800 g mol−1, and PDI of 1.03, determined by GPC (Fig. 9). The molecular weight determined by GPC was close to the theoretical value of Mn, 3500 g mol−1, further supporting the successful synthesis of conjugate 4. The difference between the two values could be caused from the calibration of column by PMMA standards. A slight tailing can be seen in the GPC traces for 4 and 5; we ascribe this to interactions between the N-alkyl urea peptoid and the stationary phase.
 | ||
| Fig. 9 GPC trace of PEG polymers showing (a) GPC trace of PEG conjugate 4; (b) GPC trace of PEG conjugate 5; (c) GPC trace of commercial PEG methyl ether tosylate; (d) flow rate marker. | ||
Although the DMB groups did not prevent the self-assembly of N-alkyl urea peptoid oligomer or the previously reported poly(N-alkyl urea peptoid),50 self-assembly did not occur for the polymer conjugate 4. Indeed, no fibers were observed to form from solutions of conjugate 4 in a variety of commonly used solvents, and NOE experiments did not show any through-space coupling peaks. These observations are unsurprising considering the hydrophilic nature of the PEG chains and resulting poor solvent compatibility with organic solvents.
We removed the DMB groups from conjugate 4 by stirring with dilute trifluoroacetic acid for 1 h at ambient temperature, making a water soluble conjugate that contained two secondary-urea functional groups (Fig. 8). This PEGylated conjugate is analogous to PEGylation of oligomer 3. The resulting PEG conjugate 5 was precipitated from cold ether with Mn of 3500 g mol−1 and a PDI of 1.03 determined by GPC using the same conditions as for conjugate 4. As expected, conjugate 5 eluted slightly later than conjugate 4 in the GPC chromatogram. The structure of conjugate 5 was confirmed using 1H NMR spectroscopy by observation of the complete disappearance of the DMB group signals (Figure S12). We prepared a 10 mg ml−1 solution of polymer conjugate 5 in deionized water and allowed the solution rest for 24 h. After this time we observed fibers suspended in the solution, examination of the fibers with SEM in an environmental mode revealed the fibers were shaped like ribbons, and were ∼ 40 μm long with a diameter of 1.1 μm (Fig. 10). This indicates that the self-assembly ability of the N-alkyl urea peptoids is retained after conjugation with PEG.
 | ||
| Fig. 10 SEM image of fibers resulting from the self-assembly of conjugate 5 in water. | ||
Conclusions
We have shown that N-alkyl urea peptoids oligomers that were observed to form fibers when polymerized appear to be undergoing intermolecular organization at concentrations below that where fiber formation occurs, and that π–π interactions do not appear crucial for intermolecular assembly. The self-assembly abilities of N-alkyl urea peptoid oligomers and polymers was extended with a novel PEGylated N-alkyl urea peptoid conjugate, and the self-assembly in aqueous environments is retained. PEGylated/N-alkyl urea peptoid conjugates represent totally synthetic analogues to polymer/peptide conjugates through using peptidomimetics oligomers. Considering the vast diversity afforded by the N-alkyl urea peptoid synthesis, this opens up possibilities in a variety of areas for these types of macromolecules, including designing biocompatible materials.Experimental
Materials and methods
All starting reagents were purchased from Aldrich at the highest purity available and used as received unless stated otherwise. The synthetic details of oligomer 1 are available in our recent paper.501H and 13C NMR measurements were recorded in CDCl3, CD3COCD3, and DMSO(d6) with Si(CH3)4, and D2O with 3-(trimethylsilyl)propionic 2,2,3,3,-d4 acid, sodium salt as internal standards using a Bruker Ultrashield 400 MHz (100 MHz for 13C); 1H NMR spectra were processed using UXNMR version 2.5 and MestRe-C. Nuclear Overhauser and Exchanged Spectroscopy (NOESY) were performed in CD2Cl2 or (d6)DMSO using a Bruker Ultrashield 500 MHz. The spectra were processed by SpinWorks 3.0 vision. Fourier transform infrared (FT-IR) spectra were collected on a Nicolet 6700 spectrometer and analyzed with OMNIC 32 software. UV-Vis absorbance spectra were collected using Cary 50 UV-VIS Absorbance Spectrophotometers. Mass spectrometry was performed using a Micromass Q-TOF-2™ spectrometer. Scanning electron microscopy (SEM) investigations were carried out on a XL30-ESEM instrument operating at energy of 20 and 30 kEV. Molecular weights of the PEG-N-alkyl urea peptoid conjugates were determined by gel permeation chromatography (GPC) using an Agilent 1100 Series HPLC equipped with a DMF mobile phase and Optilab rEX differential refractometer (light source = 658 nm) (Wyatt Technology Corporation) detector calibrated against PMMA standards (MW range = 850–1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 916000 g mol−1).
916000 g mol−1).
Synthesis of compound 2
Compound 1 (0.20 g, 0.15 x10−3 moles), and K2CO3 (41.4 mg, 0.30 x10−3 moles) in 1 mL of DMF was added to a flask and sealed with a rubber septum, purged with N2 for 30 min, and benzene thiol (68.0 mg, 6.2 x10−3 moles) added by syringe. The reaction was allowed to proceed at room temperature overnight. The DMF was removed by vacuum distillation and the residue dissolved in CH2Cl2 and passed through a thin layer of Al2O3. The solvent was removed to afford the crude secondary amine which was purified using column chromatography on basic Al2O3 with CH2Cl2 as the mobile phase. The solvent was removed and the product dried in vacuum. Yield: 0.12 g, 84%. 1H NMR (CDCl3): δ (ppm) 2.24 (s, 4 H, 2 NH2), 2.91 (s, 12 H, 4 NCH3), 3.21–3.37 (m, 28 H, NCH2 on backbone + 2 NCH2Ar), 3.81 (s, 12 H, 4 OCH3), 4.29 (s, 4 H, 2 CH2NCH2Ar), 6.12–6.74 (m, 10 H, 2 OCH3-CCHC-OCH3 + 2 CHCHC-OCH3 + 6 NHCO), 7.00–7.02 (m, 2 H, 2 CHCHC-OCH3); 13C{1H} NMR (CDCl3, 100 MHz): δ (ppm) 34.79 (2 NCH3), 34.83 (2 NCH3), 34.98 (2 CH2NH2), 39.44 (2 CH2CH2NH2), 39.47 (2 CH2CH2NCH3), 39.65 (2 CH2CH2NCH3), 45.27 (2 NCH2Ar), 45.36 (2 CH2NCH2Ar), 48.43 (2 CH2CH2NCH3), 48.50 (2 CH2CH2NCH3), 55.34 (2 OC H3), 55.43 (2 OC H3), 98.54 (2 OCH3-CCHC-OCH3), 104.34 (2 CHCHC-OCH3), 118.07 (2 CHCCH2N), 129.69 (2 CHCCH2N), 158.06 (2 CHC-OCH3), 159.27 (2 CHC-OCH3), 159.38 (2 CONH), 160.53 (2 CONH), 161.95 (2 CONH); FT-IR: (cm−1) ν(NH) = 3306, ν(alkenes) = 2933, ν(CO) = 1612, ν(Phenyls) = 1537;MS (TOF MS ES+): 933.55 (calculated: 933.56) M + 1.Synthesis of compound 3
In a 10 mL round bottom flask, 100 mg of compound 2 was dissolved in 2 mL of a solution of trifluoroacetic acid/H2O = 95/5 v/v and stirred at room temperature for 1 h. TFA was removed by purging with N2 and the residue was washed with chloroform, followed by adding 1 mL of ammonium hydroxide. The solution was stirred briefly then the residue was purified using column chromatography on basic Al2O3 with ethyl acetate/MeOH (90:10) as the mobile phase to afford compound 3. Yield: 43 mg, 53%. 1H NMR (D2O, 400 MHz): δ (ppm) 2.88 (s, 6 H, 2 NCH3), 2.91 (s, 6 H, 2 NCH3), 3.13 (t, J = 5.8 Hz, 6 H, 2 CH2NH2), 3.19 (s, 4 H, 2 NH2), 3.25–3.36 (m, 20 H, NCH2 on backbone), 3.47 (t, J = 5.6 Hz, 6 H, 2 CH2CH2NH2), 7.24 (s, appeared 2 H, NHCO exchange with D2O); 13C{1H} NMR (D2O, 100 MHz): δ (ppm) 34.44 (2 NCH3), 34.78 (2 NCH3), 37.97 (2 NCH2), 38.06 (2 NCH2), 38.57 (2 NCH2), 39.97 (2 NCH2), 40.19 (2 NCH2), 48.29 (2 CH2NCH3), 48.61 (2 CH2NCH3); FT-IR: (cm−1) ν(NH) = 3307, 1435, ν(alkenes) = 2935, ν(CO) = 1673; MS (TOF MS ES+): 633.45 (calculated: 633.76) M + 1.
Synthesis of PEG-N-alkyl urea peptoid conjugates 4
Oligomer 1 (100 mg, 0.077 x10−3 mol) was dissolved in 2 mL of N,N-dimethylformamide (DMF) and K2CO3 (21 mg, 15.4 x10−3 moles) and PEG methyl ether tosylate (170 mg, 15.4 x10−3 moles) added. The reaction was allowed to proceed at 60 °C for 48 h. The DMF was removed by vacuum distillation and the residue dissolved in CHCl3 and passed through celite. The solution was concentrated and the product isolated by column chromatography using silica gel (silica gel 60 Å, 70–230 mesh) with CH2Cl2/MeOH = 10:1 (v/v) as the mobile phase. The resulting polymer conjugate was precipitated in ice cold ether. Then the powder obtained was dried in vacuum. Yield: 150 mg, 67%. Mn of the conjugate was determined by GPC as 3700 g/mol.
Deprotection of PEG-N-alkyl urea peptoid conjugates 4 to make conjugate 5
A 10 mL round bottom flask was charged with conjugate 4 (70 mg, 0.019 x10−3 mols) and 2 mL of TFA/CH2Cl2(v/v = 1:1). The reaction flask was sealed with a rubber septum and stirred for 1 h at ambient temperature. The pink solution was dried under an air flow and redissolved in 5 ml of methanol and passed through a thin layer of celite. The resulting solution was concentrated and the final water soluble PEG conjugate was precipitated from ice cold ether. Isolated yield: 50 mg, 78%.
Sample preparation for SEM
NOESY spectra
Oligomer 1 was dissolved in CD2Cl2 in a sealed NMR tube. The full NOESY spectrum of oligomer 1 in CD2Cl2 is shown in Fig. S2a (ESI†).References
- J. F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M.v. Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- J. Hentschel and H. G. Börner, Macromol. Biosci., 2009, 9, 187–194 CrossRef CAS.
- N. Yamaguchi, B. S. Chae, L. Zhang, K. L. Kiick and E. M. Furst, Biomacromolecules, 2005, 6, 1931–1940 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C. W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- J. C. M. van Hest, Polym. Rev., 2007, 47, 63–92 CrossRef CAS.
- J. Hentschel, M. G. J. ten Cate and H. G. Börner, Macromolecules, 2007, 40, 9224–9232 CrossRef CAS.
- K. L. Heredia, L. Tao, G. N. Grover and H. D. Maynard, Polym. Chem., 2010, 1, 168–170 RSC.
- H. G. Börner, B. M. Smarsly, J. Hentschel, A. Rank, R. Schubert, Y. Geng, D. E. Discher, T. Hellweg and A. Brandt, Macromolecules, 2008, 41, 1430–143 CrossRef.
- S. Kessel and H. G. Börner, Macromol. Rapid Commun., 2008, 29, 316–320 CrossRef CAS.
- L. Huang, R. A. McMillan, R. P. Apkarian, B. Pourdeyhimi, V. P. Conticello and E. L. Chaikof, Macromolecules, 2000, 33, 2989–2997 CrossRef CAS.
- N. Yamaguchi and K. L. Kiick, Biomacromolecules, 2005, 6, 1921–1930 CrossRef CAS.
- Y. S. Jo, F. Gantz, J. A. Hubbell and M. P. Lutolf, Soft Matter, 2009, 5, 440–446 RSC.
- J. Yang, C. Xu, P. Kopečková and J. Kopeček, Macromol. Biosci., 2006, 6, 201–209 CrossRef CAS.
- S. K. Hamilton, A. L. Sims, J. Donavan and E. Harth, Polym. Chem., 2011, 2, 441–446 RSC.
- A. E. Ende, V. Sathiyakumar, R. Diaz, D. E. Hallahan and E. Harth, Polym. Chem., 2010, 1, 93–96 RSC.
- A. E. Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- J. K. Oh, S. A. Bencherif and K. Matyjaszewski, Polymer, 2009, 50, 4407–4423 CrossRef CAS.
- A. A. Dünne, H. G. Börner, H. Kukula, H. Schlaad, J. A. Werner, S. Wiegand and M. Antonietti, Anticancer Res, 2007, 27, 3935–3940 Search PubMed.
- M. Licciardi, Y. Tang, N. C. Billingham and S. P. Armes, Biomacromolecules, 2005, 6, 1085 CrossRef CAS.
- G. W. M. Vandermeulen, C. Tziatzios and H. A. Klok, Macromolecules, 2003, 36, 4107–4114 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CrossRef CAS.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- H. Li, A. P. Bapat, M. Li and B. S. Sumerlin, Polym. Chem., 2011, 2, 323–327 RSC.
- M. Li, H. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354–359 CrossRef CAS.
- S. Dehn, R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Rev., 2011, 51, 214–234 CrossRef CAS.
- H. Kakwere, R. J. Payne, K. A. Jolliffe and S. Perrier, Soft Matter, 2011, 7, 3754–3757 RSC.
- S. R. S. Ting, E. H. Min, P. Escal, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- S. A. Bencherif, N. R. Washburn and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2499–2507 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- L. Fischer and G. Guichard, Org. Biomol. Chem., 2010, 8, 3101–3117 CAS.
- L. Fischer, C. Didierjean, F. Jolibois, V. Semetey, J. M. Lozano, J.-P. Briand, M. Marraud, R. Poteau and G. Guichard, Org. Biomol. Chem., 2008, 6, 2596–2610 CAS.
- G. Guichard, A. Violette, G. Chassaing and E. Miclet, Magn. Reson. Chem., 2008, 46, 918–924 CrossRef CAS.
- C. Hemmerlin, M. Marraud, D. Rognan, R. Graff, V. Semetey, J.-P. Briand and G. Guichard, Helv. Chim. Acta, 2002, 85, 3692–3711 CrossRef CAS.
- V. Semetey, D. Rognan, C. Hemmerlin, R. Graff, J.-P. Briand, M. Marraud and G. Guichard, Angew. Chem., Int. Ed., 2002, 41, 1893–1895 CrossRef CAS.
- A. Violette, M. C. Averlant-Petit, V. Semetey, C. Hemmerlin, R. Casimir, R. Graff, M. Marraud, J.-P. Briand, D. Rognan and G. Guichard, J. Am. Chem. Soc., 2005, 127, 2156–2164 CrossRef CAS.
- A. Violette, N. Lancelot, A. Poschalko, M. Piotto, J.-P. Briand, J. Raya, K. Elbayed, A. Bianco and G. Guichard, Chem.–Eur. J., 2008, 14, 3874–3882 CrossRef CAS.
- L. Fischer, P. Claudon, N. Pendem, E. Miclet, C. Didierjean, E. Ennifar and G. Guichard, Angew. Chem., Int. Ed., 2009, 49, 1067–1070 CrossRef.
- G. Guichard, V. Semetey, M. Rodriguez and J.-P. Briand, Tetrahedron Lett., 2000, 41, 1553–1557 CrossRef CAS.
- P. Claudon, A. Violette, K. Lamour, M. Decossas, S. Fournel, B. Heurtault, J. Godet, Y. Mely, B. Jamart-Gregoire, M.-C. Averlant-Petit, J.-P. Briand, G. Duportail, H. Monteil and G. Guichard, Angew. Chem., Int. Ed., 2010, 49, 333–336 CAS.
- A. Violette, S. Fournel, K. Lamour, O. Chaloin, B. Frisch, J.-P. Briand, H. Monteil and G. Guichard, Chem. Biol., 2006, 13, 531–538 CrossRef CAS.
- X. Chen and N. Ayres, Macromolecules, 2010, 43, 1341–1348 CrossRef CAS.
- S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508–1524 CAS.
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714–721 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- X. Chen and N. Ayres, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3030–3037 CrossRef CAS.
- O. Khakshoor, B. Demeler and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 5558–5569 CrossRef CAS.
- S. Levin and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 13043–13048 CrossRef CAS.
- M. G. Woll, J. R. Lai, I. A. Guzei, S. J. C. Taylor, M. E. B. Smith and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 11077–11078 CrossRef CAS.
- D. Seebach, F. Hook David and A. Glattli, Biopolymers, 2006, 84, 23–37 CrossRef CAS.
- A. R. Sanford and B. Gong, Curr. Org. Chem., 2003, 7, 1649–1659 CrossRef CAS.
- M. George, G. Tan, V. T. John and R. G. Weiss, Chem.–Eur. J., 2005, 11, 3243–3254 CrossRef CAS.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS.
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Langmuir, 2009, 25, 8802–8809 CrossRef CAS.
- F. Rodríguez-Llansola, B. Escuder, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardin and W. Hayes, Chem. Commun., 2010, 46, 7960–7962 RSC.
- B. Song, H. Wei, Z. Wang, X. Zhang, M. Smet and W. Dehaen, Adv. Mater., 2007, 19, 416–420 CrossRef CAS.
- D. H. Williams and I. Flemming, Spectroscopic Methods in Organic Chemistry, McGraw-Hill, Maidenhead, 5th Edition edn, 1997 Search PubMed.
- F. M. Veronese and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 587–456 CrossRef CAS.
- S. Kessel, A. Thomas and H. G. Börner, Angew. Chem., Int. Ed., 2007, 46, 9023–9026 CrossRef CAS.
- H. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2009, 48, 6431–6434 CrossRef.
- T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: SEM images of the fibers formed from compound 1 and 3 with measurement of their diameters; NOESY spectra of compound 1 at different concentrations and in different solvents; UV-Vis absorption spectrum of compound 1 at a various concentration; 13C NMR and MS spectra for compounds 2 and 3 and SEM images obtained from self-assembly of conjugate 4 and 5. See DOI: 10.1039/c1py00284h/ |
| This journal is © The Royal Society of Chemistry 2011 |