Synthesis of hydroxypropyl cellulose derivatives modified with amphiphilic diblock copolymer side-chains for the slow release of volatile molecules†
Damien Loïc
Berthier
*,
Andreas
Herrmann
and
Lahoussine
Ouali
Firmenich SA, Division Recherche et Développement, B. P. 239, CH-1211 Genève 8, Switzerland. E-mail: damien.berthier@firmenich.com
First published on 8th July 2011
Abstract
Hydroxypropyl cellulose (HPC) was used as carrier material to prepare comb-like grafted block copolymers having an amphiphilic core–shell structure. The hydroxyl groups of HPC were partially protected with trimethylsilyl groups and the free OH groups used to initiate the ring opening polymerisation (ROP) of L-lactide. The end-chain of the grafted hydrophobic poly(L-lactide) (PLLA) chain was then functionalised with 2-bromopropionyl bromide which was used to polymerise the tert-butyl acrylate (tBA) monomer by atom transfer radical polymerisation (ATRP). The hydrophobic PtBA block was then hydrolysed to a hydrophilic poly(acrylic acid) (PAA) block. The resulting amphiphilic grafted copolymer HPC-g-(PLLA-b-PAA) was dissolved in an ethanol/water mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), such as a typical Eau de Toilette (EdT) formulation. Atomic force microscopy (AFM) and size measurements confirmed the presence of core–shell structures in solution. A significant swelling of HPC-g-(PLLA-b-PAA) in the presence of a volatile bioactive molecule, methyl 2,2-dimethyl-6-methylene-1-cyclohexanecarboxylate (Romascone®), was observed by dynamic light scattering (DLS). Furthermore, the presence of HPC-g-(PLLA-b-PAA) significantly decreased the evaporation rate of Romascone®, thus serving as an efficient fragrance delivery system in formulations with high ethanol content.
:1), such as a typical Eau de Toilette (EdT) formulation. Atomic force microscopy (AFM) and size measurements confirmed the presence of core–shell structures in solution. A significant swelling of HPC-g-(PLLA-b-PAA) in the presence of a volatile bioactive molecule, methyl 2,2-dimethyl-6-methylene-1-cyclohexanecarboxylate (Romascone®), was observed by dynamic light scattering (DLS). Furthermore, the presence of HPC-g-(PLLA-b-PAA) significantly decreased the evaporation rate of Romascone®, thus serving as an efficient fragrance delivery system in formulations with high ethanol content.
Introduction
Bioactive volatile compounds play a vital role in nature. As signalling compounds, they serve for the communication between plants and insects1,2 and, as a consequence of their pleasant smell, some of them are used as fragrances in our everyday life. To fulfill their biological role, these compounds have to be efficiently evaporated from various surfaces and transported through the air before reaching their target. Therefore, bioactive volatiles are typically characterised by high vapour pressures (volatilities), which at the same time limit the duration of their perception over time. To overcome this limitation, delivery systems based on chemical3,4 or physical4–6 interactions have been developed for the controlled release of fragrances. Chemical delivery systems, so-called profragrances, release volatiles from non-volatile precursor molecules by cleavage of a covalent bond,3 whereas physical delivery systems are frequently based on microcapsules which extend the stability and the long-lastingness of these volatile molecules over a certain period of time by diffusion through a shell or out of a polymer matrix.5–7In addition to microcapsules, amphiphilic block copolymers have been investigated to encapsulate drugs or volatile molecules into copolymer micelles.8–11Copolymer micelles have a higher thermodynamic stability and a better capacity to load bioactive molecules than micelles of molecular surfactants.10,12,13 Volatile molecules can be encapsulated into the cores of copolymer micelles, which was found to enhance their perception,10,14,15 as described for micelles of poly(ethylene glycol) and poly(propylene glycol) triblock copolymers in water.14 The evaporation of the volatile molecules was reduced because the micelles decreased the vapour pressure of volatile molecules when favourable intermolecular physical interactions were present in the core.14,15
Nevertheless, the formation of block copolymer micelles was found to be concentration dependent and only observed above the critical micellar concentration (cmc).16 The presence of ethanol, a solvent used in many perfumery applications, considerably modified the performance of delivery systems such as micelles of block copolymers or microcapsules.17,18 Addition of ethanol to water changed the physico-chemical parameters of the solvent mixture.19–21 On the one hand, the capsules became porous and, as a consequence, the entrapped volatile molecules leaked very rapidly into the solvent phase. On the other hand, the amphiphilic character of the block copolymers decreased and ethanol was distributed between the hydrophobic core of the micelles and the continuous phase.21 Furthermore, the cmc increased with ethanol content and micelles were usually observed only at low ethanol concentrations in water, typically below 40%.18,22 As a consequence, the controlled release of volatile molecules in self-assembled block copolymers in the presence of ethanol is extremely difficult.18
Previous work has shown that cross-linking of micelles can prevent disaggregation upon dilution or variation of composition, pH or temperature13,23–25 Nonetheless, on the basis of the large variety of chemical functions of perfumery ingredients,17 chemical cross-linking may not be appropriate. Alternatively, copolymers forming amphiphilic unimolecular micelles have been investigated17,26–39 because they exhibit micellar behaviour independent of the copolymer concentration or the composition of the solvent mixture. These amphiphilic core–shell copolymers can have a reverse or conventional micellar structure capable of encapsulating hydrophilic26–28 or hydrophobic17,29–37 molecules.
Unimolecular micelles with amphiphilic core–shell structures were prepared by using the “grafting from” method with different controlled living polymerisation processes such as ring opening polymerisation (ROP),40–42 atom transfer radical polymerisation (ATRP)43,44 or reversible addition-fragmentation transfer polymerisation (RAFT).45 Various multifunctional polymers were used to initiate polymerisation such as polythreitol,27 polyglycerol,28,29 hyperbranched polyester Boltorn® HBP H40,31,37,45,46 poly-α,β-(N-(2-hydroxyethyl)-L-aspartamide),47–49 or cellulose derivatives.13,44 Cellulose is an abundant and sustainable natural material with good biocompatibility32,50 and thus an interesting carrier for the development of polymer delivery systems. In contrast to cellulose, hydroxypropyl cellulose (HPC) is soluble in water and in organic solvents51 and was therefore chosen as a polymer backbone for the present work. Amphiphilic grafted copolymers were previously obtained from HPC.30,32,50–53 The hydroxyl groups54 can, after suitable modification (e.g. with trimethylsilyl (TMS) groups), initiate the polymerisation of biodegradable ε-caprolactone30,32,36,51,55 or lactide47,48,56,57 by ROP,40–42 or vinyl monomers by ATRP.30,44
We were interested in grafting an amphiphilic diblock copolymer onto a HPC backbone to obtain a three-dimensional carrier structure with a hydrophobic inner core (to encapsulate hydrophobic guest molecules) and a hydrophilic outer shell (to enable dispersion in aqueous media) for the slow (extended) release of bioactive compounds. We thus prepared modified HPC by consecutive grafting of a poly(L-lactide) (PLLA) and a poly(acrylic acid) (PAA) block to give a comb-like copolymer with an amphiphilic core–shell structure.58Methyl 2,2-dimethyl-6-methylene-1-cyclohexanecarboxylate (Romascone®) was used as a model fragrance to investigate the potential of the copolymer as a delivery system for the encapsulation and slow release of bioactive volatiles in ethanolic solutions.
Experimental
General
tert-Butyl acrylate (tBA) was passed through a prepacked column to remove hydroquinone and monomethyl ether hydroquinone before use. All other commercially available reagents and solvents were used without further purification. HPC with Mw = 100000 g mol−1 and a degree of polymerisation (DPn) of ca. 300 (as indicated by the manufacturer) was used for the derivatisations. Absolute ethanol had a purity of 99.8% (as indicated by the manufacturer). Reactions were carried out in standard glassware under N2. Yields were not optimised. Demineralised H2O was obtained from a Millipore-Synergy-185 water purifier. IR spectra were recorded on a Perkin-Elmer-1600-FTIR spectrometer; ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) of weak (w), medium (m), or strong (s) bands are given in cm−1. 1H and 13C NMR spectra were measured on a Bruker-DPX-400 spectrometer; δ is expressed in parts per million (ppm) downfield from Me4Si as internal standard, J is in Hz. All percentages are weight percent (wt%) relative to the total weight of the formulation. Yields of functionalisation were determined by NMR spectroscopy.
of weak (w), medium (m), or strong (s) bands are given in cm−1. 1H and 13C NMR spectra were measured on a Bruker-DPX-400 spectrometer; δ is expressed in parts per million (ppm) downfield from Me4Si as internal standard, J is in Hz. All percentages are weight percent (wt%) relative to the total weight of the formulation. Yields of functionalisation were determined by NMR spectroscopy.
Analytical size-exclusion chromatography (SEC)
SEC analyses were carried out at room temperature (r.t. ca. 22 °C) with a system composed of a Thermo Finnigan-Surveyor vacuum online degasser, quaternary LC pump, autosampler and UV/VIS detector, combined with a Thermo Separation Products Spectra-System-IR-150 refractometer and a Viscotek-270-Dual-Detector viscometer. Samples were eluted from a Macherey-Nagel Nucleogel 104-5 column (300 × 7.7 mm i.d., particle size 5 μm) at a flow rate of 1.0 mL min−1 by using HPLC-grade tetrahydrofuran (THF). Universal calibrations were performed with the viscometer and the refractive index detector using commercial poly(styrene) standards. The polymer standard (ca. 40 mg) was accurately weighed and dissolved in THF (10 mL); then those solutions (50 μL) were injected for the calibration.Preparation of polymer 1 (HPC-TMS)
HPC (4.00 g, 0.04 mmol, estimated nOH = 36 mmol) was dissolved in dry acetonitrile (MeCN; distilled and stored on molecular sieves, 60 mL). A solution of hexamethyldisilazane (HDMS, 2.66 mL, 12.8 mmol) in MeCN (10 mL) was added dropwise and the reaction mixture was heated at 90 °C for 4 h. The polymer was obtained by precipitation into water (three times) to give a white solid, which was dried under vacuum for 24 h (4.75 g, 80%). 1H NMR (CDCl3): δ 5.00–3.00 (m, 7H, HPC), 1.13 (br s, 3H, HPC), 0.20 ppm (s, 3H, TMS). 13C NMR (CDCl3): δ 102.29 (q), 82.82 (d), 81.70 (d), 75.10 (d), 74.73 (d), 73.29 (d), 67.79 (d), 67.63 (d), 68.05 to 64.81 (d), 18.56 (q), 17.99 (q), 17.41 (d), 17.34 (d), 0.27 ppm (d). FTIR (neat):max 3478 (s), 2965 (m), 2932 (m), 2896 (m), 2871 (m), 1638 (s), 1451 (s), 1406 (s), 1372 (m), 1315 (s), 1248 (s), 1083 (m), 1007 (m), 928 (s), 889 (s), 835 (s), 747 cm−1 (m). SEC: Mn 62000 g mol−1, Mw 186000 g mol−1.
Preparation of copolymer 2 (HPC-g-(PLLA)-OH)
Polymer 1 (0.20 g, 1.0 mmol) and L-lactide (7.06 g, 49.0 mmol) were dried under vacuum for 1 h and then dissolved in dry xylene (25 mL, previously distilled over CaH2). A catalytic amount of tin(II) 2-ethylhexanoate (Sn(Oct)2) was added and the reaction mixture was stirred at 135 °C for 72 h. The viscosity increased and the resulting solution was diluted in THF. The polymer was precipitated into cold n-heptane. In order to remove the remaining monomers, we dissolved the filtered solid in acetone and precipitated it into cold water. The polymer was dried under vacuum for 24 h at 50 °C to give a white solid (5.92 g, 82%). 1H NMR (CDCl3): δ 5.16 (q, J = 6.8 Hz, 26H, CH PLLA), 5.0–3.0 (m, 5H, HPC), 4.35 (q, J = 7.0 Hz, 1H, (CH3)CH-OH), 1.58 (d, J = 6.9 Hz, 78H, CH3 PLLA), 1.49 (d, J = 6.9 Hz, 3H, (CH3)CH-OH), 1.23 (m, 3H), 1.12 (m, 6H), 0.14 ppm (s, 3H). 13C NMR (CDCl3): δ 169.62 (s), 69.02 (d), 66.71 (d), 20.53 (q), 16.66 ppm (d). FTIR (neat):max 3641 (w), 3509 (w), 2996 (w), 2945 (w), 2881 (w), 1748 (s), 1453 (m), 1382 (m), 1358 (m), 1302 (w), 1267 (w), 1209 (m), 1180 (s), 1128 (s), 1085 (s), 1043 (s), 956 (w), 918 (w), 870 (m), 754 (m), 694 cm−1 (w). SEC: Mn 526000 g mol−1, Mw 810000 g mol−1.
Preparation of copolymer 3 (HPC-g-(PLLA)-Br)
Copolymer 2 (5.80 g, 0.01 mmol, estimated nOH = 7.0 mmol) was dissolved in distilled THF (20 mL). This solution was cooled to 0 °C. Then, a solution of 2-bromopropionyl bromide (1.53 g, 7.1 mmol) in THF (10 mL) was added dropwise, followed by triethylamine (Et3N, 0.71 g, 7.1 mmol). The reaction mixture was stirred at r.t. for 48 h. The polymer was precipitated into water to give a solid, which was dissolved in acetone and eventually precipitated into cold n-heptane (two times) to give a white solid (5.80 g, 92%). 1H NMR (CDCl3): δ 5.18 (q, J = 7.3 Hz, 26H, CH PLLA), 4.42 (q, J = 7.2 Hz, 1H, CH-O-CO), 5.09–2.60 (m, 5H), 1.95 (m, 3H, (CH3)CH-Br), 1.58 ppm (d, J = 7.1 Hz, 78H, CH3 PLLA). 13C NMR (CDCl3): δ 169.62 (s), 69.02 (d), 69.46 (d), 21.60 (q), 16.66 ppm (q). FTIR (neat):max 3504 (w), 2996 (w), 2945 (w), 2876 (w), 2645 (w), 1748 (s), 1724 (w), 1601 (w), 1452 (m), 1383 (m), 1358 (m), 1305 (w), 1267 (w), 1210 (m), 1180 (s), 1128 (s), 1082 (s), 1042 (s), 956 (w), 918 (w), 870 (m), 755 (m), 694 cm−1 (m). SEC: Mn 463000 g mol−1, Mw 795000 g mol−1.
Preparation of copolymer 4 (HPC-g-(PLLA)-b-(PtBA))
Copolymer 3 (1.00 g, estimated nBr = 0.7 mmol) was dissolved in tBA (15.54 mL, 107.0 mmol) in the presence of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 148 μL, 0.7 mmol). The oxygen of the reaction mixture was removed by three consecutive freeze–pump–thaw cycles before CuBr was added (0.10 g, 0.7 mmol). The reaction mixture was stirred at 100 °C for 30 min. After freezing into liquid nitrogen to stop the polymerisation, the reaction mixture was dissolved in chloroform. The organic phase was washed with an aqueous solution of HCl (0.01 N), and then the organic layer was dried (MgSO4), filtered and concentrated. The polymer was precipitated into a methanol/water (9:1, v/v) mixture to give a white solid, which was dried under vacuum for 24 h (3.11 g, 68%). 1H NMR (CDCl3): δ 5.16 (q, J = 7.0 Hz, 1H, CH PLLA), 2.50 (m, 3.5H), 1.58 (d, J = 7.1, 3H, CH3 PLLA), 1.44 (m, 35H, (CH3)3C), 1.27 ppm (m, 4.5H). 13C NMR (CDCl3): δ 174.19 (s), 173.99 (s), 169.62 (s), 80.34 (s), 69.01 (d), 42.41 (d), 41.91 (d), 41.58 (d), 37.43 (t), 36.03 (t), 28.10 (q), 16.66 ppm (q). FTIR (neat): max 2975 (m), 2931 (m), 2873 (w), 1757 (m), 1721 (s), 1478 (m), 1449 (m), 1391 (m), 1365 (s), 1252 (s), 1216 (m), 1180 (s), 1141 (s), 1085 (s), 1042 (m), 910 (w), 844 (s), 752 cm−1 (m). SEC: Mn 704000 g mol−1, Mw 1810000 g mol−1.
Preparation of copolymer 5 (HPC-g-(PLLA)-b-(PAA))
Copolymer 4 (1.00 g, estimated ntBA = 4.26 mmol) was dissolved in dichloromethane (10 mL). Trifluoroacetic acid (TFA, 10 mL) was added and the reaction mixture was stirred at r.t. for 1 h to give a suspension. The polymer was eventually precipitated into cold diethyl ether to give a white solid (0.61 g, 93%). 1H NMR (MeOD): δ 5.19 (q, J = 7.0 Hz, 1H, CH PLLA), 2.44 (m, 6H, CH PAA), 1.95–1.71 (m, 10H, CH2 PAA), 1.55 ppm (d, J = 7.0 Hz, 3H, CH3 PLLA). 13C NMR (MeOD): δ 178.51 (s), 171.03 (s), 70.45 (d), 42.93 (s), 42.72 (s), 42.43 (s), 37.22 (t), 36.35 (q), 35.67 (q), 17.06 ppm (q). FTIR (neat):max broad peak from 3686 to 2147 (m), 2938 (m), 1754 (s), 1697 (s), 1450 (m), 1413 (m), 1382 (m), 1361 (m), 1159 (s), 1086 (s), 1045 (s), 796 cm−1 (s). SEC: No data were recorded in THF or in water.
Sample preparation for release experiments
Samples were prepared by direct addition of the copolymer to different solvents at r.t. (T = 20 °C) and mixing with a magnetic stir bar overnight. Samples without perfume were prepared to measure the size of the polymer particle in water, ethanol/water mixtures (9:1 wt/wt), and n-butyl acetate. For Romascone®-containing samples, the volatile was added to the copolymer solution and then stirred overnight as described earlier. These samples contained 10% of volatile molecule, 85% of ethanol/water mixture (9:1) and 5% of polymer.
Viscosity and refractive index measurements
Viscosity measurements were carried out with a Viscolite 700 viscometer from Hydramotion. Solutions were introduced in a water bath at 20 °C. Densities were measured at 20 °C with a DMA 4500 densimeter from Anton Paar. Refractive indices were measured at 20 °C with a DR6200T refractometer from A. Krüss Optronic GmbH.Dynamic light scattering (DLS)
DLS measurements were performed at 20 °C with a Zetasizer, Nanoseries, Nano-ZS apparatus (Malvern Instruments) equipped with a 4 mW He–Ne laser at a wavelength of 633 nm. Data were analysed using the CUMULANTS analysis mode to determine the hydrodynamic diameter, expressed as the mean size of particles (Zave) and the polydispersity index (Ip).Atomic force microscopy (AFM)
AFM topographic and phase images of individual core–shell copolymer 5 were obtained using a Multimode Atomic Force Microscope (Veeco Metrology Group) equipped with a Nanoscope IV control station and a 5695JV scanner in tapping mode. Pointprobe®-Silicon SPM-Sensor cantilevers (Nanoworld, thickness 4 μm, length 125 μm, width 30 μm) with a resonance frequency of 320 kHz and a force constant of 42 N m−1 were used. The samples were prepared by deposition of 1 μL of solution of 5 at 0.02% in an ethanol/water mixture (9:1) onto freshly cleaved mica. Multiple images from different areas of the sample were collected to obtain an accurate representation of the molecule.
Thermogravimetric analysis (TGA)
Taking into account the high volatilities of the compounds in the sample (ethanol, water and perfume molecule), the solutions (10 μL) were injected with a precision syringe (Hamilton, 10 μL) into an aluminium crucible (Mettler-Toledo ME-26763, 40 μL) previously weighed on a precision balance (Mettler-Toledo XP204S). The densities of the solutions were not affected by the addition of copolymers, and the corresponding masses weighed on the balance, before TGA, were not impacted by this modification of the density. The crucibles were capped during the transfer from the balance to the TGA autosampler (Mettler-Toledo TSO801RO). Samples were analysed under a constant nitrogen flow of 20 mL min−1 with a TGA/SDTA851ethermogravimetric analyser (Mettler-Toledo) equipped with a microbalance (accuracy: 1 μg) and an accurate oven having an internal volume of 35 mL. The elapsed time between the injection and the first point recorded by the TGA cannot be controlled accurately and was estimated at between 2 and 3 min. The temperature profile started at 25 °C for 1 min, was then ramped to 50 °C at 5 °C min−1, and kept at 50 °C for 25 min, to result in a total run time of 31 min. Measurements were performed in triplicate.Results and discussion
Synthesis of copolymers
Polysaccharides are typically silylated with HDMS or chlorotrimethylsilane (TMSCl)56,57,59 to control the number of OH groups and, as a consequence, the degree of substitution on the polysaccharide backbone. In general, HDMS is preferred over TMSCl, because the latter releases HCl, which might degrade the polysaccharide backbone.56 HDMS reacts with two OH groups to give two TMS groups. From previous work on the silylation of HPC with HDMS,59 we decided to target a TMS/OH molar ratio of 1:3 to prepare TMS-functionalised polymer HPC-TMS (1), as illustrated in Scheme 1. We decided to work with this molar ratio to have on the one hand a dense substitution on the polysaccharide backbone and on the other hand to prevent any cross-linking during the ATRP step. Knowing that the conversion is not complete (we typically found conversions around 50%), we used a two-fold excess of HDMS for the reaction. The yield of the reaction (80%) was then determined from the ratio of the weight of isolated polymer 1 and the corresponding theoretical weight expected for the monofunctionalisation with HDMS.
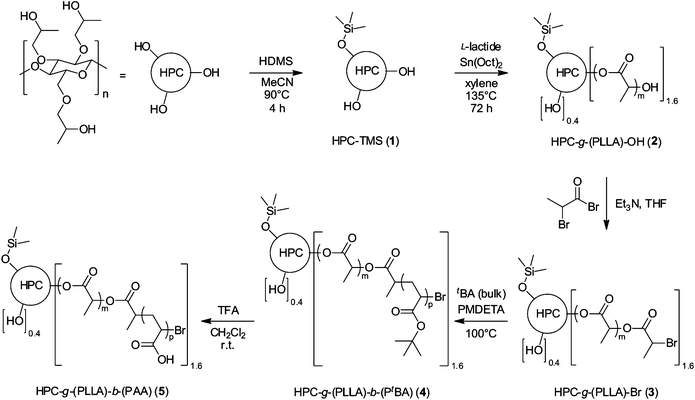 | ||
| Scheme 1 Preparation of polymers 1–5 from HPC (arbitrary distribution of the side-chains with respect to the HPC backbone). | ||
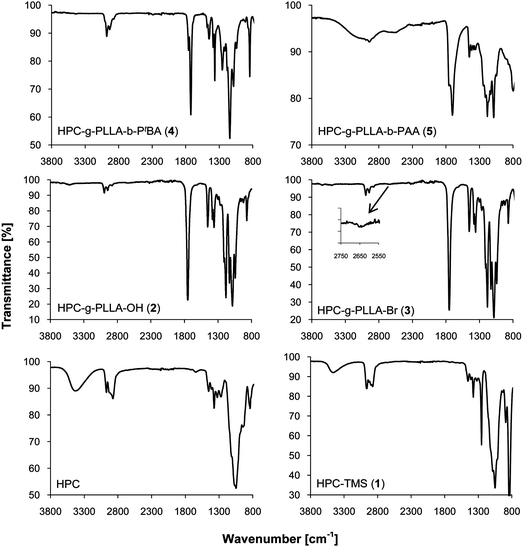
FTIR analysis of 1 showed peaks at 1248, 889, 835 and 747 cm−1, all corresponding to the TMS groups (Fig. 1). As a result of partial substitution, the remaining OH groups gave a broad signal at 3478 cm−1. The degree of substitution was determined by NMR spectroscopy in CDCl3 to be 33% by integrating the signals of the TMS groups at 0.20 ppm and of the methyl protons of HPC at 1.12 ppm (Fig. 2). The degree of substitution is in agreement with the selected initial molar ratio, resulting in two free OH groups per repeating unit in 1.
 | ||
| Fig. 1 Comparison of the FTIR of polymers 1–5. | ||

Grafted cellulose derivatives have previously been constructed with vinylic monomers by using a Ce(IV)-based redox process for the polymerisation.60 To prepare biodegradable cellulose materials, we decided not to build up the hydrophobic part of the core–shell structure with vinyl monomers but with a biodegradable PLLA unit which can be achieved by ROP of L-lactide. Polymerisation was thus initiated on the free OH groups of the polysaccharide, as previously described for the reaction of ε-caprolactone.30,61 Homogeneous ROP of L-lactide was carried out in xylene at 135 °C, in the presence of tin(II) 2-ethylhexanoate as catalyst, to prepare poly(L-lactic acid)-grafted HPC copolymer 2 (HPC-g-(PLLA)-OH) (Scheme 1).
FTIR analysis showed new peaks from the PLLA at 2995 and 2945 cm−1 arising from the alkyl chain and at 1753 cm−1 corresponding to the carbonyl group (Fig. 1). A weak peak observed at 3510 cm−1 was attributed to the stretching of the remaining OH functions. NMR analysis of 2 in CDCl3 showed signals from the PLLA block, with a doublet at 1.58 ppm for the methyl group and a quadruplet at 5.16 ppm for the methyne group (Fig. 2). In contrast, the doublet at 1.49 and the quadruplet at 4.35 ppm were attributed to the last PLLA unit of the chain. The average DPn of the PLLA block was estimated at 26 by comparing the integrations of the signals at 5.16 and 4.35 ppm (Fig. 2). Zoom A of Fig. 2 shows a weak signal at 3.5 ppm, which was attributed to the CH and CH2 group of the HPC backbone. The average degree of grafting per HPC unit was determined by integration of the signal corresponding to the proton of the central HPC unit (between 3 and 5 ppm) and the signal of the methyne groups from the PLLA chain (at 5.16 ppm). Another possibility to determine the degree of functionalisation would be to integrate NMR signals resulting from the TMS groups. We have not chosen this option due to the weak intensity of this signal in the 1H NMR spectrum and possible overlap of this peak with that of the Me4Si (or its satellites) used as internal standard for the NMR measurements. The methyl groups of the PLLA chain with a DPn of 26 give rise to a signal intensity corresponding to 78 protons. If all hydroxyl groups of the HPC unit were substituted, one would expect a signal intensity of 3.5 for the protons of the CH–O and CH2–O groups of the HPC. In our case, this signal integrated for 5 protons, thus suggesting a degree of functionalisation of approximately 1.6. As previously observed for a similar case,37 it should be noted that with increasing size of the core–shell structure, it is becoming more and more difficult to identify (and therefore to quantify) NMR signals arising from the core of the copolymer.
Average molecular weights of HPC and polymers 1–4 were measured by SEC in THF (Table 1 and Fig. 3). The molecular weight of polymer 1 decreased as compared with the value of the HPC starting material. Because molecular weights were determined by viscosity measurements and because the presence of TMS groups increased the solubility of polymer 1 in THF, the viscosity of the solution decreased and, as a consequence of changes in the conformation of the polymer chains with respect to HPC, thus resulted in a decrease in Mw. The Mn of copolymer 2 was 526000 g mol−1 (Table 1), thus corresponding to a DPn of 16 for the PLLA block. This value is below the one determined by NMR, which is in agreement with the generally observed tendency that SEC typically underestimates the molecular weights of grafted block copolymers as compared with their linear equivalents.62–64 The Ip was determined to be 1.54, as compared with a value of 3.02 which was measured for polymer 1. This difference might be explained by the presence of a large number of PLLA chains being grafted onto 1. These chains have a narrow polydispersity and thus lower the difference between Mn and Mw for copolymer 2.
| Polymer | M n/g mol−1 | M w/g mol−1 | I p |
|---|---|---|---|
| HPC | 82000 |
298000 |
3.63 |
| 1 | 62000 |
186000 |
3.02 |
| 2 | 526000 |
810000 |
1.54 |
| 3 | 463000 |
795000 |
1.72 |
| 4 | 704000 |
1810000 |
2.57 |
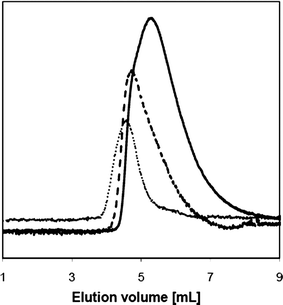 | ||
Fig. 3
Size exclusion chromatography of polymers 1 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), 3 ( ), 3 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and 4 (…) in THF at r.t. ) and 4 (…) in THF at r.t. | ||
The polymerisation of tBA by ATRP requires the presence of an alkyl halide function such as an α-halo bromide. The preparation of a poly(L-lactic acid)-grafted HPC copolymer with a terminal bromide function (HPC-g-(PLLA)-Br, 3) was carried out by esterification of the ω-hydroxyl function of the PLLA block with 2-bromopropionyl bromide in the presence of Et3N to give a terminal alkyl bromide function on this unit (Scheme 1).65
1H NMR analysis of 3 showed a shift of the quadruplet corresponding to the OH at the chain end of the PLLA block from 4.36 to 4.42 ppm, which indicated quantitative esterification (Zoom B in Fig. 2). At the same time, a new singlet for the methyl group of the 2-bromopropionyl bromide was observed. The presence of the 2-bromopropionyl bromide was confirmed by 13C NMR analyses (see the ESI†). The doublet corresponding to the last CH group of the PLLA chain was shifted from 66.71 ppm (alcohol) to 69.02 (ester), and the signal of the terminal CH3 group was shifted from 20.53 to 21.60 ppm.‡ Furthermore, FTIR spectroscopy showed the presence of two new weak peaks at 1724 (as a weak shoulder in the peak from PLLA ester groups) and 2645 cm−1 (zoom in Fig. 1), the former corresponding to the stretching of the carbonyl of the ester group, the latter to the stretching of the carbon–bromide bond (Fig. 1). Finally, with a measured Mn of 463000 g mol−1 (Table 1 and Fig. 3), SEC analysis showed a modest change of the average molecular weights.
Grafted HBC diblock copolymer HPC-g-(PLLA)-b-(PtBA) (4) with a PLLA and a PtBA block was obtained by polymerisation of tBA by ATRP, using copolymer 3 as the initiator (Scheme 1). This reaction was performed in bulk in the presence of a large excess of the tBA monomer. Copper (I) bromide with PMDETA as ligand was used as the polymerisation catalyst. A high concentration of polymers with terminal bromide functions has been reported to generate high concentrations of radicals during propagation, which then causes cross-linking between the polymer chains.66–72 Despite low concentrations of radicals during propagation in the ATRP process, the methacrylate monomer was found to be too reactive. The viscosity increased during the polymerisation to eventually give a gel. To avoid this phenomenon, we decided to work with less reactive tBA in bulk.67–69 Previous work showed that low conversions avoided gelification of the reaction mixture.67–69,71 As a consequence, we used high monomer/initiator molar ratios (see Experimental part) to obtain a minimum conversion.67
Nevertheless, the viscosity of the solution increased very rapidly and the reaction was stopped after 30 min at a conversion of 35%. This is presumably due to the high density of initiating bromide groups on the copolymer surface. The 1H NMR signal at 1.43 ppm, corresponding to the tert-butyl group of the polyacrylate block, was compared with the signal at 5.16 ppm, corresponding to the CH group of the PLLA block. The DPn of PLLA was 26, whereas the DPn of the PtBA block was determined by NMR spectroscopy to correspond to 100 units (Fig. 2). SEC measurements in THF gave an average Mw of about 1.8 × 106 g mol−1 (Table 1).
Finally, to obtain amphiphilic HPC copolymer HPC-g-(PLLA)-b-(PAA) (5) with a PLLA and a PtBA block, the tert-butyl ester of the PtBA block was hydrolysed with TFA to give a PAA block.28,30Copolymer 4 was dissolved in dichloromethane in the presence of a large excess of TFA (Scheme 1). Copolymer 5 started to precipitate after 1 h and was isolated by filtration and rinsing with diethyl ether. The resulting copolymer was analysed by NMR spectroscopy in methanol (Fig. 4). Peaks from the polyacrylate backbone were observed at 1.83 and 2.46 ppm, whereas the peak at 1.43 ppm from the PtBA group disappeared. FTIR analysis showed a very broad peak from 3740 to 2350 cm−1 of the carboxylic acid and two strong peaks at 1700 and 1750 cm−1 for the carbonyl groups of the acid and the PLLA, respectively (Fig. 1). These results indicated complete hydrolysis of the PtBA block to PAA.
Release of Romascone®
HPC itself has amphiphilic properties, and we observed that it is thermodynamically compatible with bioactive volatile molecules such as fragrances. A solution of HPC (10%, Mw = 100000 g mol−1) in Romascone® gave a gel which might behave as a diffusion-controlled delivery system. As the basis of this work, we assumed that HPC-g-(PLLA)-b-(PAA) (5), consisting of a HPC backbone grafted with a hydrophobic block of PLLA and a hydrophilic block of PAA, may act as a core–shell delivery system for volatile molecules, as observed recently for hyperbranched polymer-based core–shell structures37,46,72 or self-assembled block copolymer micelles.18 The hydrophobic core of HPC and PLLA should function as a nanocontainer for hydrophobic molecules.30 Furthermore, the hydrophilic shell was expected to favour the formation of dispersions in water or ethanol and thus slow down the diffusion of hydrophobic volatile molecules through the hydrophilic PAA shell as a result of low thermodynamic compatibility. As outlined in the Introduction, the presence of ethanol strongly influences the physico-chemical parameters of the system. Particularly challenging are Eau de Toilette (EdT) formulations with a total ethanol content of more than 75%. Previous investigations carried out with star block copolymers17 showed that the polymers were not soluble at an ethanol concentration above 60%. On the other hand, micelles of Pluronic copolymers were disaggregated in solutions containing more than 40% of ethanol.18
The particular advantage of copolymer 5 is its complete solubility in ethanol/water mixtures with high ethanol content. We expected copolymer 5 to adopt a core–shell configuration in an EdT, as previously observed for other core–shell systems30,37,72 or microcapsules73 in media with much lower ethanol content or even without ethanol. Copolymer 5 was dissolved at 0.2% in an EdT formulation which typically consists of an ethanol/water mixture (9:1) containing 10% of volatile molecules. A clear solution was obtained. Size measurements carried out by DLS showed the formation of particles with an average diameter of 64.5 nm, whereas unmodified HPC displayed an average diameter of 27.2 nm under the same conditions (Fig. 5 and Table 2).
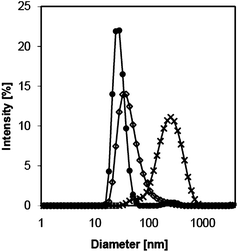 | ||
| Fig. 5 Size measurements of HPC (●) and copolymer 5 (◊) in an ethanol/water mixture (9:1) and of copolymer 5 in an EdT formulation in the presence of Romascone® (×), measured at 20 °C by DLS. | ||
The observation of globular structures by AFM suggested the presence of core–shell systems. A solution of 5 (at 0.02% in the ethanol/water mixture) was used to obtain a monomolecular film on mica (Fig. 6). The AFM topographic and phase images clearly show the individual globular structure of 5 with a diameter measured at 56 nm, in agreement with the mean diameter determined by DLS. The slight decrease in size measured by AFM with respect to the DLS analyses might be explained by the absence of solvent in the former case.

The presence of a hydrophobic core should allow entrapment of volatile hydrophobic molecules and the slowing down of their release in an EdT. In the context of this work, we studied the evaporation of Romascone® as a model fragrance molecule. Size measurements indicated that copolymer 5 significantly swelled in the EdT formulation to give a structure with an average diameter of about 203 nm, which was twice as large as the diameter measured in the pure solvent mixture (Fig. 5 and Table 2). Ethanol and water being unsuitable solvents for PLLA, we assumed that the observed swelling was due to the entrapment of Romascone® in the core of the copolymer. In the presence of water and ethanol, hydrophobic Romascone® should preferentially interact with the PLLA block of the copolymer, as observed for hyperbranched polymer-based core–shell structures37,46,72 or micelles of Pluronic copolymers.18
The determination of the precise loading capacity (e.g. by diffusion NMR spectroscopy)72 turned out to be difficult. We supposed this might be due to the high Romascone® content which changed the physico-chemical properties of the solvent with respect to the copolymer solubility. In addition to the observed swelling, this could induce a change in size distribution as a result of the dynamic self-assembly of the copolymer aggregates. The addition of the copolymer was expected to reduce the chemical potential and to influence the vapour pressure of volatile molecules, as described in the Flory mean-field approximation.17 The efficiency of a delivery system can therefore be easily estimated by measuring the weight loss resulting from the evaporation of the volatile molecule in the presence or absence of the copolymer. A lower evaporation rate of the volatile then corresponds to increased performance.17,73TGA is a convenient method to measure this weight loss as a function of time in a dynamic process.17,18,73
TGA of the solvent mixture showed a complete evaporation after 4.2 min at 50 °C (Fig. 7). Under the same conditions, the evaporation of Romascone® (at 10%) from the earlier described EdT formulation was monitored in the presence and absence of copolymer 5. Fig. 7 shows that the ethanol/water solvent mixture evaporated quite rapidly within 7 min (Part A). Only 0.46 mg of the original solution remained after this initial period, presumably being pure Romascone®. The observed weight loss stopped completely after 12.8 min when all the Romascone® was evaporated (Part B). The average evaporation rate calculated for the reference solution between 7 and 12.8 min was estimated to be 63.9 μg min−1. In the presence of copolymer 5 (at 5%), the measured weight loss lasted up to 25.4 min, compared with the 12.8 min observed in the experiment without copolymer. The evaporation rate was estimated at 42.8 μg min−1, which is significantly lower than that of the reference.
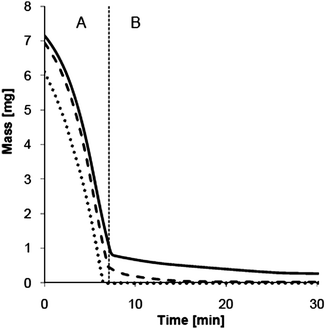 | ||
| Fig. 7
TGA of ethanol/water 9:1 (…) and Eau de Toilette formulations containing 10% of Romascone® as a reference () and of Romascone® in the presence of copolymer 5 () at 50 °C for 30 min. | ||
In contrast to previous delivery systems,17,18DLS measurements and AFM images indicate that incorporation of Romascone® in the core of 5 is possible despite the high concentration of ethanol in the solution. As a consequence, addition of 5% of copolymer 5 decreased the evaporation rate of Romascone® from 63.9 to 42.8 μg min−1 and significantly improved the long-lastingness of this volatile molecule in the investigated EdT application.
Conclusions
The development of efficient fragrance delivery systems in media with high ethanol content (such as EdT formulations) is challenging. Previously developed core–shell structures failed in these applications, as the polymers were not soluble at high ethanol concentrations, while copolymer micelles typically disaggregated in these media. Amphiphilic comb-like copolymers with an HPC backbone, an inner hydrophobic PLLA and an outer hydrophilic PAA block were found to be a suitable fragrance delivery system for applications with high ethanol content. The copolymers were prepared in a five-step sequence from HPC by partial silylation of the hydroxyl groups, ROP of L-lactide to build the hydrophobic shell and functionalising of the outer surface to serve as macroinitiator for ATRP. Polymerisation of tBA and hydrolysis of the tert-butyl group finally afforded HPC-g-PLLA-b-PAA (5). In the presence of a large amount of ethanol, this copolymer preserved its core–shell structure, as demonstrated by DLS and AFM measurements. In the presence of Romascone® as a bioactive volatile molecule, copolymer 5 swelled in an ethanol/water mixture with ethanol concentrations above 75% to give particles with an average diameter of ca. 200 nm. The influence of 5 on the evaporation profile of Romascone® was assessed by TGA at 50 °C. The measured weight loss of the EdT solution showed that the evaporation rate of Romascone® was significantly reduced in the presence of the copolymer, thus giving rise to the desired long-lastingness of fragrance perception. In application, tailor-made, biodegradable amphiphilic core–shell structures with different hydrophobic and hydrophilic blocks have a strong potential to efficiently control the evaporation of various volatile ingredients. Further investigations will focus on a better understanding of the nature of the different blocks to influence the release properties of volatile compounds, and of mixtures of volatiles, with different functionalities.Notes and references
- See, for example, The Chemistry and Biology of Volatiles, ed. A. Herrmann, John Wiley & Sons, Chichester, 2010 Search PubMed; L. M. Schoonhoven, J. J. A. van Loon and M. Dicke, Insect–Plant Biology, Oxford University Press, Oxford, 2nd edn, 2005 Search PubMed; Plant–Animal Interactions: an Evolutionary Approach, ed. C. M. Herrera and O. Pellmyr, Blackwell Publishing, Oxford, 2002 Search PubMed.
- See, for example, Special section on plant volatiles, Science, 2006, 311, 803–819 Search PubMed.
- A. Herrmann, Angew. Chem., Int. Ed., 2007, 46, 5836–5863 CrossRef CAS; A. Herrmann, in The Chemistry and Biology of Volatiles, ed. A. Herrmann, John Wiley & Sons, Chichester, 2010, pp. 333–362 Search PubMed.
- J. Karlsen, in Handbook of Essential Oils, ed. K. H. C. Baser and G. Buchbauer, CRC Press, Boca Raton, 2010, pp. 855–861 Search PubMed.
- J. Ness, O. Simonsen and K. Symes, Microspheres, Microcapsules Liposomes, 2003, 6, 199–234 CAS.
- S.-J. Park and R. Arshady, Microspheres, Microcapsules Liposomes, 2003, 6, 157–198 CAS.
- Delivery System Handbook for Personal Care and Cosmetic Products, ed. M. R. Rosen, William Andrew Inc., Norwich, 2005 Search PubMed.
- K. Kataoka, G. S. Kwon, M. Yokoyama, T. Okano and Y. Sakurai, J. Controlled Release, 1993, 24, 119–132 CrossRef CAS.
- A. V. Kabanov, I. R. Nazarova, I. V. Astafieva, E. V. Batrakova, V. Y. Alakhov, A. A. Yaroslavov and V. A. Kabanov, Macromolecules, 1995, 28, 2303–2314 CrossRef CAS.
- Y. Saito, K. Miura, Y. Tokuoka, Y. Kondo, M. Abe and T. Sato, J. Dispersion Sci. Technol., 1996, 17, 567–576 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS.
- T. Satoh, Soft Matter, 2009, 5, 1972–1982 RSC.
- K. Suzuki, Y. Saito, Y. Tokuoka, M. Abe and T. Sato, J. Am. Oil Chem. Soc., 1997, 74, 55–59 CrossRef CAS.
- S. E. Friberg, Q. Yin and P. A. Aikens, Colloids Surf., A, 1999, 159, 17–30 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- C. Ternat, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco, C. J. G. Plummer, G. Kreutzer, H.-A. Klok, J.-A. E. Månson and A. Herrmann, Macromolecules, 2008, 41, 7079–7089 CrossRef CAS.
- D. L. Berthier, I. Schmidt, W. Fieber, C. Schatz, A. Furrer, K. Wong and S. Lecommandoux, Langmuir, 2010, 26, 7953–7961 CrossRef CAS.
- P. Alexandridis and L. Yang, Macromolecules, 2000, 33, 5574–5587 CrossRef CAS.
- R. Ivanova, B. Lindman and P. Alexandridis, Langmuir, 2000, 16, 3660–3675 CrossRef CAS.
- P. Alexandridis, R. Ivanova and B. Lindman, Langmuir, 2000, 16, 3676–3689 CrossRef CAS.
- C. Chaibundit, N. M. P. S. Ricardo, F. M. L. L. de Costa, M. G. P. Wong, D. Hermida-Merino, J. Rodriguez-Perez, I. W. Hamley, S. G. Yeates and C. Booth, Langmuir, 2008, 24, 12260–12266 CrossRef CAS.
- H. Huang, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1999, 121, 3805–3806 CrossRef CAS.
- K. B. Thurmond, II, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240 CrossRef.
- Q. Ma and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4805–4820 CrossRef CAS.
- K. Kono, T. Fukui, T. Takagishi, S. Sakurai and C. Kojima, Polymer, 2008, 49, 2832–2838 CrossRef CAS.
- Y. Kitajyo, Y. Nawa, M. Tamaki, H. Tani, K. Takahashi, H. Kaga, T. Satoh and T. Kakuchi, Polymer, 2007, 48, 4683–4690 CrossRef CAS.
- D. Kul, L. M. Van Renterghem, M. A. R. Meier, S. Strandman, H. Tenhu, S. S. Yilmaz, U. S. Schubert and F. E. Du Prez, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 650–660 CrossRef CAS.
- J. Han and C. Gao, Curr. Org. Chem., 2011, 15, 2–26 CrossRef CAS.
- E. Östmark, D. Nyström and E. Malmström, Macromolecules, 2008, 41, 4405–4415 CrossRef.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 3009–3019 CrossRef CAS.
- M.-F. Hsieh, N. V. Cuong, C.-H. Chen, Y. T. Chen and J. M. Yeh, J. Nanosci. Nanotechnol., 2008, 8, 2362–2368 CrossRef CAS.
- R. K. Kainthan, C. Mugabe, H. M. Burt and D. E. Brooks, Biomacromolecules, 2008, 9, 886–895 CrossRef CAS.
- H. Liu, S. Farrell and K. Uhrich, J. Controlled Release, 2000, 68, 167–174 CrossRef CAS.
- H. Liu, A. Jiang, J. Guo and K. E. Uhrich, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 703–711 CrossRef CAS.
- C. Xia, X. Ding, Y. Sun, H. Liu and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4013–4019 CrossRef CAS.
- C. Ternat, G. Kreutzer, C. J. G. Plummer, T. Q. Nguyen, A. Herrmann, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco, H.-A. Klok and J.-A. E. Månson, Macromol. Chem. Phys., 2007, 208, 131–145 CrossRef CAS.
- J. Bernard, M. Schappacher, P. Viville, R. Lazzaroni and A. Deffieux, Polymer, 2005, 46, 6767–6776 CrossRef CAS.
- J. Bernard, M. Schappacher, A. Deffieux, P. Viville, R. Lazzaroni, M.-H. Charles, M.-T. Charreyre and T. Delair, Bioconjugate Chem., 2006, 17, 6–14 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- D. J. A. Cameron and M. P. Shaver, Chem. Soc. Rev., 2011, 40, 1761–1776 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel and J. Bernard, Macromol. Rapid Commun., 2010, 31, 1751–1772 CrossRef CAS.
- S. Luo, C. Ling, X. Hu, X. Liu, S. Chen, M. Han and J. Xia, J. Colloid Interface Sci., 2011, 353, 76–82 CrossRef CAS.
- G. Kreutzer, C. Ternat, T. Q. Nguyen, C. J. G. Plummer, J.-A. E. Månson, V. Castelletto, I. W. Hamley, F. Sun, S. S. Sheiko, A. Herrmann, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco and H.-A. Klok, Macromolecules, 2006, 39, 4507–4516 CrossRef CAS.
- T. Peng, S.-X. Cheng and R.-X. Zhuo, J. Biomed. Mater. Res., Part A, 2006, 76, 163–173 CrossRef.
- H. J. Lee, S. R. Yang, E. J. An and J.-D. Kim, Macromolecules, 2006, 39, 4938–4940 CrossRef CAS.
- T. Peng, J. Su, S.-X. Cheng and R.-X. Zhuo, J. Biomater. Sci., Polym. Ed., 2006, 17, 941–951 CrossRef CAS.
- L. Ma, H. Kang, R. Liu and Y. Huang, Langmuir, 2010, 26, 18519–18525 CrossRef CAS.
- R. Shi and H. M. Burt, J. Appl. Polym. Sci., 2003, 89, 718–727 CrossRef CAS.
- L. Ma, R. Liu, J. Tan, D. Wang, X. Jin, H. Kang, M. Wu and Y. Huang, Langmuir, 2010, 26, 8697–8703 CrossRef CAS.
- E. Östmark, S. Harrisson, K. L. Wooley and E. E. Malmström, Biomacromolecules, 2007, 8, 1138–1148 CrossRef.
- T. Ouchi, T. Kontani, R. Aoki, T. Saito and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6402–6409 CrossRef CAS.
- P. De Leonardis, L. Mannina, M. Diociaiuti and G. Masci, Polym. Int., 2010, 59, 759–765 CAS.
- C. Nouvel, C. Frochot, V. Sadtler, P. Dubois, E. Dellacherie and J.-L. Six, Macromolecules, 2004, 37, 4981–4988 CrossRef CAS.
- Q. Cai, Y. Wan, J. Bei and S. Wang, Biomaterials, 2003, 24, 3555–3562 CrossRef CAS.
- Parts of this publication are the subject of a patent application: D. L. Berthier and L. Ouali (to Firmenich SA), WO 2005/108471, 2005.
- C. Nouvel, I. Ydens, P. Degée, P. Dubois, E. Dellacherie and J.-L. Six, Polymer, 2002, 43, 1735–1743 CrossRef CAS.
- O. Y. Mansour and A. Nagata, Prog. Polym. Sci., 1985, 11, 91–165 CrossRef CAS.
- C. Wang, Y. Dong and H. Tan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 273–280 CrossRef CAS.
- T. Kaneko, T. Horie, M. Asano, T. Aoki and E. Oikawa, Macromolecules, 1997, 30, 3118–3121 CrossRef CAS.
- H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CrossRef CAS.
- S. Förster, I. Neubert, A. D. Schlüter and P. Lindner, Macromolecules, 1999, 32, 4042–4049 Search PubMed.
- H. R. Kricheldorf, J. M. Jonté and M. Berl, Makromol. Chem., Suppl., 1985, 12, 25–38 CrossRef CAS.
- J. N. Kizhakkedathu, R. Norris-Jones and D. E. Brooks, Macromolecules, 2004, 37, 734–743 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 2000, 33, 7261–7274 CrossRef CAS.
- G. Cheng, A. Böker, M. Zhang, G. Krausch and A. H. E. Müller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- L. Bes, S. Angot, A. Limer and D. M. Haddleton, Macromolecules, 2003, 36, 2493–2499 CrossRef CAS.
- K. Ohno, B. Wong and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2206–2214 CrossRef CAS.
- W. Fieber, A. Herrmann, L. Ouali, M. I. Velazco, G. Kreutzer, H.-A. Klok, C. Ternat, C. J. G. Plummer, J.-A. E. Månson and H. Sommer, Macromolecules, 2007, 40, 5372–5378 CrossRef CAS.
- L. Ouali, G. León, V. Normand, H. Johnsen, A. Dyrli, R. Schmid and D. Benczédi, Polym. Adv. Technol., 2006, 17, 45–52 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1py00195g |
| ‡ Please note that the 2-bromopropionyl bromide might also react with the OH groups of the HPC backbone. This reaction is less likely to proceed due to the steric hindrance of the grafted PLLA chains. |
| This journal is © The Royal Society of Chemistry 2011 |