Synthesis of a linear polymer from an AB2 monomer of 1-(3-phenoxypropyl)piperidine-4-one in trifluoromethanesulfonic acid
Youko
Tamura
,
Yumiko
Ito
,
Yukari
Segawa
,
Tomoya
Higashihara
and
Mitsuru
Ueda
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1-H120, O-okayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: ueda.m.ad@m.titech.ac.jp; Fax: +81-3-5734-2127; Tel: +81-3-5734-2127
First published on 7th June 2011
Abstract
A linear polymer was successfully prepared by self-polycondensation of 1-(3-phenoxypropyl)piperidine-4-one as an AB2 monomer, which was previously reported to produce a hyperbranched polymer (HBP) with 100% degree of branching (DB). Based on results of the model reaction, the DB could be controlled by changing the acidity of polymerization media.
Introduction
Encouraging progress has been made over the last few years in the synthesis of HBPs.1–5 Conventional HBPs are prepared by one-step polymerization of ABx monomers, where A and B represent two different functional groups, and the DB of the HBPs is determined by statistics and only reaches around 50%, assuming it is based on equal reactivity of the two B functional groups of the AB2 monomer.6 The DB is controlled by the reactivity of the functional groups involved in the synthesis. 100% HBPs can be obtained when the first reaction step of an AB2 monomer activates the second one. Recently, Smet et al. reported the synthesis of HBPs with 100% DB by the polymerization of an AB2 type monomer, such as the acid-catalyzed polycondensation of isatins or acenaphthenequinones with aromatic compounds.7,8 We also reported the synthesis of a series of 100% HBPs by the acid-catalyzed polycondensation of 2-(4-phenoxyphenoxy)fluorene, 2-[4-(4-mercaptobutoxy)-9H-fluorene-9-one or 1-(3-phenoxypropyl)piperidine-4-one, and base-catalyzed polycondensation of 3,3-dibromo-1-hexyl-5-hydroxyindoline-2-one.9–12 Furthermore, a linear polymer (DB = 0) was produced from an AB2 monomer, 2,2,2-trifluoro-1-[4-(4-phenoxyphenoxy)phenyl]ethanone in TFSA.13 Quite recently, HBPs with controlled DBs from 0 to 100% have been reported by the self-polycondensation of 2,2,2-trifluoro-1-[4-(4-phenoxyphenoxy)phenyl]ethanone (TPPE) as a monomer by using different amounts of TFSA.14 These polymerizations rely on a change in the rate determination steps in the mono- and diarylation of 2,2,2-trifluoroacetophenone by changing the molar ratios of [TFSA]/[TPPE]. This finding prompted us to synthesize linear polymers from the AB2 monomers described above. Acid-catalyzed 100% HBPs were prepared in methanesulfonic acid (MSA). Thus, self-polycondensations of these monomers are expected to produce linear polymers in TFSA in place of MSA.In this paper, we present the synthesis of a linear polymer by self-polycondensation of an AB2 monomer 1-(3-phenoxypropyl)piperidine-4-one in TFSA. Moreover, a model reaction between 1-ethylpiperidine-4-one and anisole was carried out to investigate the reactivity of the second B versus that of the first of the AB2 monomers.
Experimental
Materials
All reagents were purchased from TCI Co. Ltd. (Japan) and used without further purification. N-Methyl-2-pyrrolidone (NMP) dehydrated was used as received.Measurements
The FT-IR spectra were measured on a Horiba FT-720 spectrophotometer. The 1H and 13C NMR spectra were recorded on a Bruker DPX-300 spectrometer at 300 MHz for 1H and 75 MHz for 13C measurement. Thermogravimetry (TG) was performed using a Seiko TG/DTA 6300 thermal analysis system at a heating rate of 10 °C min−1 under nitrogen.Model reaction
To a solution of 1-ethylpiperidine-4-one (1, 0.500 g, 3.93 mmol) and anisole (2, 0.425 g, 3.93 mmol) was added TFSA (4.00 mL, 45.5 mmol) and then the solution was stirred at 0 °C for 5 min. The solution was poured into anhydrous methanol. The aqueous solution was then made basic by addition of saturated NaHCO3 aq, and the products were extracted with dichloromethane. The organic solution was then washed with water and brine, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with hexane/ethyl acetate (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), to give 1-ethyl-4-(4-methoxyphenyl)-1,2,3,6-tetrahydropyridine (4, 0.75 g) in 90% yield. IR (neat, v, cm−1): 2966, 2792, 1604, 1511, 1473, 1272, 1241, 1184, 1145, 1115, 1033, 952, 825. 1H NMR (300 MHz, CDCl3, δ, ppm): 7.32 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.97 (m, 1H), 3.80 (s, 3H), 3.14 (q, J = 3.0 Hz, 2H), 2.70 (t, J = 5.6 Hz, 2H), 2.56 (m, 2H), 2.53 (q, J = 7.3 Hz, 2H), 1.16 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3, δ, ppm): 158.7, 134.3, 133.7, 125.9, 120.2, 113.7, 55.3, 52.9, 52.1, 50.2, 28.3, 12.4. Anal. Calcd for C14H19NO: C, 77.38; H, 8.81; N, 6.45. Found: C, 77.01; H, 8.69; N, 6.16.
:1), to give 1-ethyl-4-(4-methoxyphenyl)-1,2,3,6-tetrahydropyridine (4, 0.75 g) in 90% yield. IR (neat, v, cm−1): 2966, 2792, 1604, 1511, 1473, 1272, 1241, 1184, 1145, 1115, 1033, 952, 825. 1H NMR (300 MHz, CDCl3, δ, ppm): 7.32 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.97 (m, 1H), 3.80 (s, 3H), 3.14 (q, J = 3.0 Hz, 2H), 2.70 (t, J = 5.6 Hz, 2H), 2.56 (m, 2H), 2.53 (q, J = 7.3 Hz, 2H), 1.16 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3, δ, ppm): 158.7, 134.3, 133.7, 125.9, 120.2, 113.7, 55.3, 52.9, 52.1, 50.2, 28.3, 12.4. Anal. Calcd for C14H19NO: C, 77.38; H, 8.81; N, 6.45. Found: C, 77.01; H, 8.69; N, 6.16.
Synthesis of 1-(3-phenoxypropyl)piperidine-4-one (7)
Monomer 7 was prepared from piperidine-4-one hydrochloride and (4-bromobutoxy)benzene in DMF according to a previous paper.11Polymer synthesis
TFSA (4.1 mL) was added to a solution of 7 (0.02 g, 0.086 mmol) in NMP (0.1 mL) cooled with an ice bath. The solution was stirred at this temperature for 3 h, and then poured into anhydrous methanol. The aqueous solution was then made basic by addition of saturated NaHCO3 aq, and the polymer precipitated was filtered. The polymer was collected and dried at 80 °C for 8 h under vacuum to give faint yellow powder (0.019 g, 99%). IR (KBr, v, cm−1): 3043, 2946, 2773, 1639, 1604, 1511, 1473, 1380, 1249, 1180, 1029, 825, 640. 1H NMR (protonated 8) (300 MHz, (CD3)2CO, δ, ppm): 7.37 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.06 (br, 1H), 4.27 (br, 1H), 4.14 (br, 2H), 3.96 (br, 2H), 3.62 (br, 3H), 2.90 (br, 2H), 2.38 (br, 2H). 13C NMR (75 MHz, (CD3)2CO, δ, ppm): 160.0, 137.5, 132.7, 127.6, 126.6, 115.8, 115.2, 66.4, 55.0, 52.2, 50.8, 25.4. Anal. Calcd for C14H19NO 1.86H2O: C, 67.32; H, 8.96; N, 5.60. Found: C, 67.69; H, 6.75; N, 5.50.Results and discussion
Model reaction
In a previous paper,11 we reported that a diarylated piperidine, 1-ethyl-4,4-bis(4-methoxyphenyl)piperidine (3), was obtained by the reaction of 1 and an excess amount of 2 at room temperature in MSA or TFSA. Furthermore, it was found that the rate constant of the second substitution is much faster than that of the first from the model reaction of 1 and 2 in MSA (Scheme 1).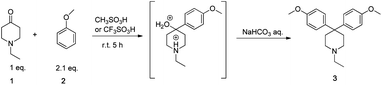 | ||
| Scheme 1 Reaction of 1 and 2 in MSA or TFSA. | ||
However, the reaction between equimolar amounts of 1 and 2 in TFSA has not been performed in detail. Thus, a model reaction between equimolar amounts of 1 and 2 was carried out in TFSA to clarify the reactivity of the second B versus that of the first of the AB2 monomers. The reaction was completed in 5 min at 0 °C and the solution was poured into anhydrous methanol (Scheme 2).
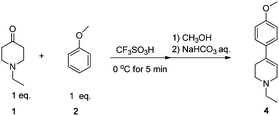 | ||
| Scheme 2 Reaction between an equimolar amount of 1 and 2 in TFSA. | ||
The structure of the isolated compound 4 was characterized based on elemental analysis as well as FT-IR, 1H and 13C NMR spectroscopy. The IR spectrum of compound 4 showed a characteristic absorption at 1630 cm−1 which is assignable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching. No absorption due to the CO group of 1 was observed at 1716 cm−1. Fig. 1 shows the 1H NMR spectrum of compound 4 with the assignment of the observed resonances. The peaks due to characteristic methoxy and olefin protons are observed at 3.80 and 5.97 ppm, respectively. In the 13C NMR spectrum of compound 4, twelve signals are observed as expected (Fig. 2).
C stretching. No absorption due to the CO group of 1 was observed at 1716 cm−1. Fig. 1 shows the 1H NMR spectrum of compound 4 with the assignment of the observed resonances. The peaks due to characteristic methoxy and olefin protons are observed at 3.80 and 5.97 ppm, respectively. In the 13C NMR spectrum of compound 4, twelve signals are observed as expected (Fig. 2).
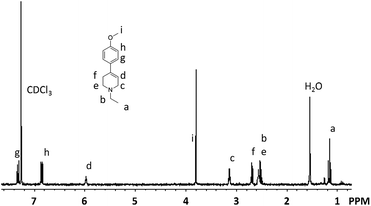 | ||
| Fig. 1 1NMR spectrum of compound 4. | ||
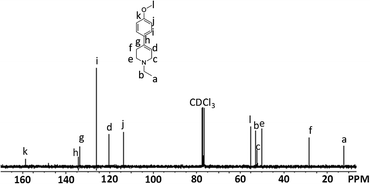 | ||
| Fig. 2 13C NMR spectrum of compound 4. | ||
The structure of 4 was further confirmed by the elemental analysis, in which the measured C, H, and N compositions agreed well with the calculated values. These observations suggested the following plausible reaction mechanism for the formation of 4. The diprotonated carbonyl compound of 1 in TFSA reacts with 2 to give intermediate 6, whose reactivity is lower than that of protonated 1 (5). Because an equivalent amount of 2 to 1 is used, no compound 2 remains in the reaction solution after the first arylation. Thus, intermediate 6 remains in TFSA after consumption of 2. If intermediate 6 is more reactive than 5, diarylated compound 3 is formed even in the presence of an equivalent of 2 to 1. On the other hand, when two equivalents of 2 to 1 are used, compound 2 attacks intermediate 6 to produce diarylated compound 3 (Scheme 3).
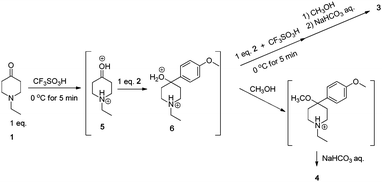 | ||
| Scheme 3 Plausible reaction mechanism. | ||
When the solution of intermediate 6 in TFSA was poured into anhydrous methanol, the product would be 1-ethyl-4-methoxy-4-(4-methoxyphenyl)piperidine, which is easily converted to 4 through elimination of methanol. To elucidate the above mechanism, after the reaction between equimolar amounts of 1 and 2 for 5 min, one equivalent of 2 was added to the reaction solution. The product was diarylated compound 3, supporting the above plausible mechanism.
Synthesis of polymers
According to the results of the experiments described above, all requirements to produce a linear polymer from an AB2 monomer are satisfied; the reactive species of 1 is formed in TFSA and then condenses with 2 to produce intermediate 6 which remains in TFSA after consumption of 2. Therefore, polymerization of 7, which produced a HBP with a DB of 100% in MSA (Scheme 4),11 was performed in TFSA. Monomer 7 was added to TFSA under various conditions as summarized in Table 1.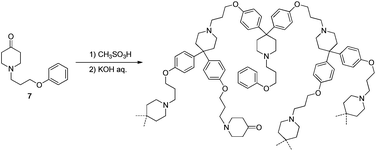 | ||
| Scheme 4 Synthesis of HBP with a DB of 100% using 7 as a monomer. | ||
| Run | Conc./mol l−1 | Temp./°C |
|---|---|---|
| a Polycondensation was carried out with 8.6 × 10−2 mmol of monomer 7 in TFSA for 5 min. | ||
| 1 | 0.10 | 0 |
| 2 | 0.10 | −5 |
| 3 | 0.10 | −22 |
| 4 | 0.17 | 0 |
| 5 | 0.020 | 0 |
| 6 | 0.020 | −5 |
| 7 | 0.020 | −22 |
Although the reaction temperature and concentration were changed, gel formation immediately occurred and isolated polymers were insoluble in organic solvents such as N,N-dimethylformamide, NMP, trifluoroacetic acid (TFA) and MSA probably because of cross-linking reactions. The viscous oily monomer takes time to dissolve in TFSA and oligomerization occurs on the surface of 7. Thus, the carbocations formed may attack the phenyl rings of other oligomers to yield cross-linked polymers. To prevent the cross-linking reaction, monomer 7 was dissolved in NMP and the solution was added to TFSA. In this case, polymerization proceeded in a homogeneous state without gelation (Scheme 5). The results of polymerizations are summarized in Table 2. The polymer was isolated by pouring the solution into anhydrous methanol. The obtained polymer was a faint yellow solid and was soluble in TFA at room temperature. Polymer 8 was characterized by FT-IR and NMR spectroscopy. In the FT-IR spectrum, the characteristic absorption of CC and ether bonds appeared at 1639 and 1249 cm−1, respectively.
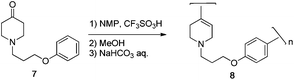 | ||
| Scheme 5 Synthesis of polymer 8. | ||
| Run | Temp./°C | Time/min | Yield (%) |
|---|---|---|---|
| a Polycondensation was carried out with 8.6 × 10−2 mmol of monomer 7 in TFSA (4.1 mL) in the presence of NMP. | |||
| 8 | −17 | 5 | 82 |
| 9 | 0 | 5 | 89 |
| 10 | 0 | 10 | 96 |
| 11 | 0 | 15 | 97 |
| 12 | 0 | 180 | 99 |
The 1H NMR spectrum of protonated 8, which is soluble in acetone-d6, is presented in Fig. 3 with the assignments of all peaks. The signals at 7.37 and 6.88 ppm assignable to aromatic protons and 6.06 to olefin protons are clearly observed. The degree of polymerization (DP) of polymer 8 was determined to be 15 by NMR spectroscopy, where the integral ratios between aromatic protoni (2 protons) and the end aromatic proton B (2 protons) in polymer 8 were used.
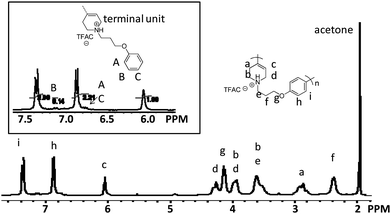 | ||
| Fig. 3 1H NMR spectrum of protonated polymer 8. | ||
Furthermore, the structure of polymer 8 was investigated by 13C NMR spectroscopy. The characteristic aromatic carbon next to the ether bond and the olefin carbon of protonated polymer 8 are observed at 160.0 and 115.8, respectively. The 13C NMR spectra of protonated compound 3, compound 4, and polymer 8 are shown in Fig. 4. No signal at 44 ppm which is assigned to the quaternary carbon of the dendritic unit was observed in the spectrum of polymer 8, which means that polymer 8 does not have a branching unit.
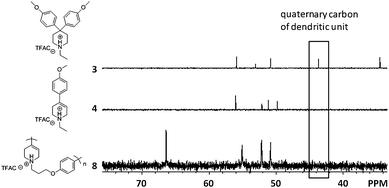 | ||
| Fig. 4 13C NMR spectra of protonated compound 3, 4 and polymer 8. | ||
The thermal properties of polymer 8 were evaluated by TG. The polymer exhibited a 5% weight loss at 260 °C in nitrogen.
Conclusions
We demonstrated that linear polymer 8 in place of the HBP with 100% DB in MSA was successfully prepared by self-polycondensation of 7 in TFSA. The results of a model reaction between equimolar amounts of 1 and 2 in TFSA clearly supported the formation of linear polymer 8. The acid strength in sulfonic acids changes the rate-determining steps in the mono- and diarylation of 1, which induces HBPs with various DBs. Therefore, these findings will accelerate the synthesis of various series of HBPs with controlled DBs and their application to functional materials.Acknowledgements
The financial support from Japan Science and Technology (JST) PRESTO program (#JY220176) in part is gratefully acknowledged. Y. S. and Y. I. thank Yoshida Scholarship Foundation and Japan Society of the Promotion of Science (JSPS, #2208196), respectively, for financial supports.References
- M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS.
- Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS.
- B. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef CAS.
- C. J. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4583 CrossRef CAS.
- M. Smet, E. H. Schacht and W. Dehaen, Angew. Chem., Int. Ed., 2002, 41, 4547 CrossRef CAS.
- Y. Fu, A. Vandendriessche, W. Dehaen and M. Smet, Macromolecules, 2006, 39, 5183 CrossRef CAS.
- S. Kono, W. Sinananwanich, Y. Shibasaki, S. Ando and M. Ueda, Polym. J., 2007, 39, 1150 Search PubMed.
- W. Sinananwanich and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2689 Search PubMed.
- W. Sinananwanich, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 994 CrossRef CAS.
- W. Sinananwanich, Y. Segawa, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 8718 Search PubMed.
- Y. Segawa, W. Sinananwanich, T. Higashihara and M. Ueda, Macromolecules, 2008, 41, 8309 Search PubMed.
- Y. Segawa, T. Higashihara and M. Ueda, J. Am. Chem. Soc., 2010, 132, 1100 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |