Catalytic synthesis and post-polymerization functionalization of conjugated polyisoprene†
William M.
Gramlich
ab and
Marc A.
Hillmyer
*a
aDepartment of Chemistry, University of Minnesota, 207 Pleasant St SE, Minneapolis, MN 55455-0431, USA. E-mail: hillmyer@umn.edu
bDepartment of Chemical Engineering and Material Science, University of Minnesota, 421 Washington Ave. SE, Minneapolis, MN 55455-0132, USA
First published on 15th June 2011
Abstract
Anionically synthesized polyisoprene (PI) was catalytically isomerized with RuHCl(CO)(PPh3)3 to produce conjugated PI (CPI). Both the reaction time and temperature were varied to control the number of conjugated dienes along the CPI backbone. The same reaction scheme also conjugated polycyclooctadiene, demonstrating the utility of the catalyst to conjugate other olefin-containing polymers. The CPI samples retained their narrow distributions as indicated by size exclusion chromatography (SEC). Small molecules were coupled with CPI through the Diels–Alder click reaction in solution to produce an array of post-polymerization functionalized polymers. In one example, N-2-hydroxyethyl maleimide (HEMI) was coupled to CPI to produce a hydroxylated material. L-Lactide was polymerized from this macroinitiator with both tin(II) octoate (Sn(Oct)2) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) to produce poly(L-lactide) (PLLA) grafted from PI (PI-g-PLLA) with a monomodal SEC distribution at 95 wt% PLLA content.
Polyisoprene (PI) has recently been produced synthetically from renewable materials using a fermentation process.1 Renewably synthesized combined with oxidative degradation characteristics, PI will be established as a sustainable polymer. The low glass transition temperature of PI makes it attractive as a component in adhesives, rubbers, and tough materials, while the chemical structure of PI (isolated carbon–carbon double bonds) allows for chemical modifications to produce a broad array of functional polymers.2–5 The renewably sourced nature and versatility of PI make it a desirable component for new materials design.
The functionalization of polymer chains is a widely used technique to create specialty polymers.6 New properties and complex architectures (star, branched, graft, etc.) are produced from functionalized polymers. Functionality may be incorporated into a polymer by copolymerization with a monomer containing the desired functionality. Many times, no such monomer exists that can be readily copolymerized at conditions compatible with its functionality, requiring the functionalization post-polymerization.6 Post-polymerization functionalization of polymers allows for one parent polymer chain to undergo many different reactions to produce an array of new functional materials from that parent polymer.
One emerging method to facilitate the post-polymerization functionalization of polymers is “click chemistry” because these click reactions are high yield, modular, and yield no side products.7–10 For these reasons, click reactions provide a versatile and efficient method to functionalize polymers post-polymerization. Common functionalizations include thiol–ene,11azide–alkyne,10 and Diels–Alder reactions.12 Among these reactions, typically, only the Diels–Alder reaction does not require a catalyst or initator.7 Diels–Alder reactions have been used to produce star polymers,13,14 block polymers,15graft copolymers,12 reversibly crosslinked polymers,16 and small molecule functionalized polymers.17–19 For PI to undergo similar Diels–Alder reactions, it must contain conjugated dienes or a reactive dienophile.
Conjugation of the double bonds on PI has been accomplished using several methods. In most cases, conducting polymers are the final goal. Perhaps the most discussed method to conjugate PI is doping the polymer with molecular iodine (I2).20 However, the I2 doped polymers do not only contain conjugated dienes and polyene segments, they also have multiple intermediates from the conjugation process such as charge complexes, cation–radical intermediates, and iodated double bonds.21–24 Other oxidants such as SbCl5 and TiCl4 have also been used to conjugate polydienes with similar results and intermediates.25,26 The aforementioned techniques to produce conjugated dienes on PI, though effective at producing conducting polymers, do not necessarily provide the functionality and stability required for the desired Diels–Alder reactions due to the high concentration of undesirable side products. Elimination of brominated poly(isobutylene-co-isoprene) has produced conjugated dienes available for Diels–Alder reactions, but at low concentrations (0.1 mmol g−1polymer).27,28 These limitations of previous approaches to PI conjugation motivate the development of new conjugation methods.
Transition metal catalysts are widely used to isomerize and migrate carbon–carbon double bonds in small molecules.29–33 Typically, these reactions conserve the original number of carbon–carbon double bonds, only moving double bonds along the carbon chain. Under appropriate conditions, the double bonds will migrate over several carbon atoms to reach the most energetically favorable position, for example adjacent to another π-orbital containing functional group.34,35 If additional carbon–carbon double bonds are present in the molecule, systems of conjugated dienes can be formed as has been demonstrated with linoleic and linolenic acids and esters.36–38 Much like the fatty acids, PI contains isolated double bonds along its chain, suggesting that PI can be conjugated with similar transition metal catalysts.
In analogy to the conjugation of vegetable oils and related fatty acids, we investigated the ability of the RuHCl(CO)(PPh3)3 catalyst to conjugate olefins along the PI backbone. We successfully synthesized conjugated PI (CPI) with varying degrees of conjugation. The conjugated dienes along CPI reacted with small molecules through the Diels–Alder click reaction to produce tailored functionality along the polymer backbone. As an example of the utility of this approach, we functionalized a sample of CPI with primary hydroxyl groups. The hydroxyl group functionalized CPI was used as the macroinitiator for the ring opening polymerization of L-lactide, creating poly(L-lactide) (PLLA) graft CPI copolymers (PI-g-PLLA). Previous work in our group has shown that similar graft copolymers of polylactide with an incompatible, low glass transition temperature polymer backbone result in materials with high toughness.39
Results and discussion
Conjugation of polyisoprene
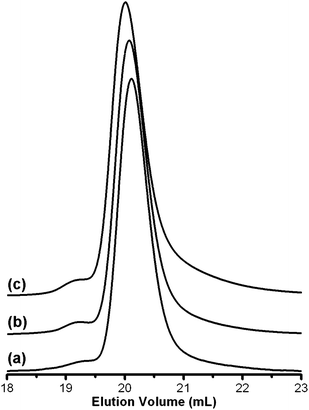
CPI was synthesized from anionically polymerized PI (see Experimental details) using RuHCl(CO)(PPh3)3 at [Ru]/[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C] = 1.6 × 10−3 in benzene (Table 1). Generally, the CPIs had similar number average molecular weights (Mn) and only slightly larger polydispersity index (PDI) values as compared to the original PI when measured by size exclusion chromatography (SEC) using polystyrene standards. The SEC traces (Fig. 1 and ESI†) of the CPIs have a higher molecular weight shoulder that is consistent with a small amount of coupling and a tail consistent with some limited degradation likely due to the increased reactivity of the conjugated products. Addition of the antioxidant butylated hydroxytoluene (BHT) to the product thwarted the coupling and degradation as shown in Fig. 1b compared to the original PI trace (Fig. 1a). Conjugation reactions were also performed on polycyclooctadiene (PCOD) to produce conjugated PCOD (CPCOD) (see ESI†) with results similar to PI, indicating that a similar reaction scheme can be used to conjugate other olefin-containing polymers.
C] = 1.6 × 10−3a
C] = 1.6 × 10−3 in benzene (Table 1). Generally, the CPIs had similar number average molecular weights (Mn) and only slightly larger polydispersity index (PDI) values as compared to the original PI when measured by size exclusion chromatography (SEC) using polystyrene standards. The SEC traces (Fig. 1 and ESI†) of the CPIs have a higher molecular weight shoulder that is consistent with a small amount of coupling and a tail consistent with some limited degradation likely due to the increased reactivity of the conjugated products. Addition of the antioxidant butylated hydroxytoluene (BHT) to the product thwarted the coupling and degradation as shown in Fig. 1b compared to the original PI trace (Fig. 1a). Conjugation reactions were also performed on polycyclooctadiene (PCOD) to produce conjugated PCOD (CPCOD) (see ESI†) with results similar to PI, indicating that a similar reaction scheme can be used to conjugate other olefin-containing polymers.
C] = 1.6 × 10−3a
| Sample designationb | Reaction time/h | Percent CC conjugatedc (%) |
M n d/kg mol−1 | PDId |
|---|---|---|---|---|
| a Concentration of the polymer in benzene was 20% w/v, BHT was added to samples after conjugation to prevent coupling and degradation. b CPI = conjugated polyisoprene, the number following is the average number of E2 dienes per CPI molecule, calculated by 1H NMR spectroscopy; Mn of PI = 25 kg mol−1. c Of all the olefins along the polymer backbone, this is the percentage that are in conjugation as calculated by 1H NMR spectroscopy. d Measured using SEC calibrated with polystyrene standards. | ||||
| PI | 50 | 1.05 | ||
| CPI-4.4 | 44 | 6 | 42 | 1.20 |
| CPI-17 | 160 | 20 | 48 | 1.08 |
| CPI-30 | 400 | 31 | 44 | 1.11 |
The proposed CPI synthesis mechanism (Fig. 2) follows those for the isomerization of small molecules.30 The reaction passes through series of steps where the ruthenium-hydride catalyst adds across a double bond and then undergoes reductive elimination. The addition and subsequent elimination can isomerize the isolated carbon–carbon double bond (e.g. E to Z conformation) or lead to migration of the bond along the polymer chain. One such migration event in PI brings carbon–carbon double bonds closer together in a bis-allylic configuration. Subsequent isomerizations/migrations to the bis-allylic intermediate result in conjugated dienes with varying stereochemistry (Fig. 2).
 | ||
| Fig. 2 Proposed isomerization mechanism of polyisoprene by the ruthenium hydride catalyst and major conjugated diene isomers present in CPI. At least two isomerization events are required to produce conjugated dienes along the polymer chain. Conjugated diene isomers are named according to the conformation of the more substituted double bond and placement of methyl group (E1, E2, and Z2). | ||
Analysis of the conjugation reaction products by proton 1H NMR spectroscopy (Fig. 3) confirmed the formation of both the intermediate bis-allylic protons (2.5–3.0 ppm) and the formation of the conjugated dienes (5.4–6.5 ppm). Other resonances at chemical shifts consistent with conjugated diene formation are also present (see ESI† for detailed assignments) as well as peaks corresponding to the original PI. Three major conjugated diene stereoisomers were identified (Fig. 2): E1, E2, and Z2. The E2 and Z2 isomers make up the bulk of the conjugated dienes in nearly equal proportion, while the E1 isomer comprises the remaining 5–10% of the conjugated dienes.
 | ||
| Fig. 3 Representative 1H NMR spectra of CPI with selected peak assignments. Expanded sections of spectra show peaks consistent with conjugated olefinic protons and bis-allylic protons. The asterisk marks the 3,4 isomer of PI present in the sample. See ESI† for more detailed assignments. | ||
Reaction time, temperature, and catalyst loading were varied to investigate the degree of conjugation attainable (Table 1 and ESI†). The percent of carbon–carbon double bonds conjugated increased from 6 to 31% with an increase in reaction time from 44 to 400 h at 60 °C (Table 1). The long times required for higher degrees of conjugation at 60 °C are likely due to the low catalyst loading (1.6 × 10−3 [Ru]/[CC]). Typically, isomerization reactions on small molecules with RuHCl(CO)(PPh3)3 at similar temperatures and in solvent are performed at catalyst loadings ten times greater than used to conjugate PI and reach completion in a matter of hours.34,40,41 Addition of more catalyst to the system was limited by its solubility in benzene (see ESI†). Increasing the reaction temperature (75–120 °C) in an effort to increase the reaction rate gave mixed results. In general, at temperatures higher than 60 °C the initial conjugation rate increased, but over time the degrees of conjugation were below that achievable by running the reaction at 60 °C (see ESI†).
Diels–Alder reactions with CPI
To functionalize PI post-conjugation, we investigated the reactions of small molecule dienophiles (R) and end functionalized PLLA (see ESI†) with select CPIs. To probe the susceptibility of the CPI to a Diels–Alder reaction, the small molecules we studied varied in “dienophilicity” (Table 2). Analysis of an initial coupling reaction between N-2-hydroxyethyl maleimide (HEMI) and CPI by 1H NMR spectroscopy (see ESI†) indicated that the HEMI predominately reacted with the E2 isomer of CPI. The methyl groups in the E1 and Z2 conjugated dienes presumably hinder the cycloaddition reaction (see ESI†). Conversely, the E2 isomer (Fig. 4) is relatively unhindered and consequently more reactive.42 All subsequent Diels–Alder reactions with HEMI and other small molecules favored reaction with the E2 isomer of CPI. Similar small molecule reactions also were observed with CPCOD (see ESI†).| Ra | [R]/[E2]b | Reaction temperature/°C | Reaction time/h | Grafts per polymerc | M n d/kg mol−1 | PDId |
|---|---|---|---|---|---|---|
| a Small molecule to be grafted to the polymer; HEMI = N-2-hydroxyethyl maleimide, MA = maleic anhydride, HEA = 2-hydroxyethyl acrylate, HEMA = 2-hydroxyethyl methacrylate. b Ratio of moles of small molecule (R) to moles of E2 CPI isomer. c The number of small molecule grafts to the polymer, found from 1H NMR spectroscopy. d Calculated from SEC calibrated by polystyrene standards. | ||||||
| HEMI | 1.0 | 110 | 20 | 30 | 49 | 1.23 |
| MA | 1.1 | 110 | 20 | 33 | 44 | 1.16 |
| HEA | 1.0 | 160 | 111 | 26 | 40 | 1.24 |
| HEMA | 1.0 | 160 | 111 | 8 | 29 | 1.38 |
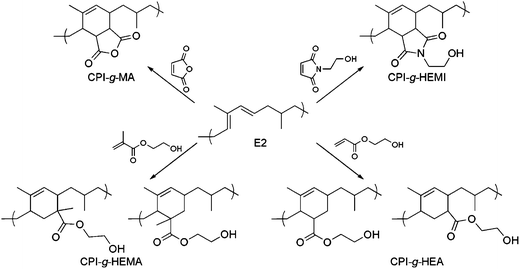 | ||
| Fig. 4 Proposed chemical structures of products from small molecules reacting with CPI. The conformation required for the Diels–Alder cycloaddition is shown for the E2 isomer of CPI. While CPI-g-HEMI and CPI-g-MA form one regioisomer, the Diels–Alder products with HEMA and HEA form two regioisomers. 1H-NMR spectra of products with peak assignments can be found in the ESI†. | ||
Both HEMI and maleic anhydride (MA) coupled with CPI-30 (Fig. 4) to near completion in 20 h (110 °C) when loaded at a [HEMI or MA]/[E2] = 1. SEC analysis of the grafted products (Fig. 1c) indicates that grafting at 110 °C does not significantly change the molecular weight distribution compared to the original CPI. Similar reactions between conjugated PCOD and both HEMI and MA resulted in complete conversion as well (see ESI†). At the same reaction conditions, the poorer dienophiles vinyl acetate (VA), 2-hydroxyethyl acrylate (HEA), 2-hydroxyethyl methacrylate (HEMA), and 1-vinyl-2-pyrrolidone (V2P) did not graft onto the CPI backbone. Increasing the reaction temperature to 160 °C and time to 111 h resulted in the productive reaction between HEA and HEMA with the E2 isomer to form CPI-g-HEA and CPI-g-HEMA respectively (Fig. 4). The apparent decrease in reactivity of HEMA compared to HEA as evidenced by the degree that each coupled to CPI-30 is likely due to the extra methyl group of HEMA increasing the bulkiness of the molecule. VA and V2P still did not react with CPI at 160 °C for 115 h, presumably due to the electron donating groups present in each molecule.42 Increasing the reaction time and temperature allowed for HEMA and HEA to graft, but consequently the PDI of the CPI broadened and the Mn decreased significantly. SEC traces of the products (see ESI†) have features that are both consistent with CPI degradation and coupling caused by thermally induced free radical reactions.
Synthesis of CPI-g-PLLA
CPI-17 functionalized with 19 HEMI molecules was used as the macroinitiator for CPI-g-PLLA synthesis with a target of 95 wt% PLLA. Three widely used lactide polymerization catalysts were investigated: tin(II) octoate (Sn(Oct)2), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), and AlEt3. The AlEt3 polymerization led to a product with a multimodal SEC distribution (see ESI†) and low conversion. Conversely, the Sn(Oct)2 and TBD catalyzed reactions produced graft copolymers with monomodal distributions (Fig. 5) and PDI values of 1.08 and 1.11 respectively. The shift to lower elution volume in the SEC traces is consistent with graft copolymer formation. A small broad peak exists in the SEC trace of the Sn(Oct)2 synthesized CPI-g-PLLA which may correspond to a low level of PLLA homopolymer. In both the SEC traces for the Sn(Oct)2 and TBD catalyzed polymerizations, the peak associated with the macroinitiator is absent, suggesting that all or nearly all of the macroinitiator reacted. | ||
| Fig. 5 SEC elution curves of (a) CPI-g-HEMI macroinitiator and CPI-g-PLLA synthesized using (b) Sn(Oct)2 and (c) TBD catalysts respectively. Shift to lower elution volume is consistent with graft copolymer formation. | ||
1H NMR spectroscopy was used to confirm the synthesis of CPI-g-PLLA (Fig. 6) from CPI-g-HEMI. Peaks associated with the methylene protons of HEMI grafted from CPI (Fig. 6a) shifted downfield with the formation of PLLA. Peaks consistent with the PLLA end groups (4.4 ppm and 2.6 ppm) as well as the PLLA repeat units appeared confirming the formation of PLLA. The integration ratios of the HEMI and PLLA end groups agree with expectations for graft copolymer formation and complete initiation. The Mn of the individual PLLA grafts were calculated by end group analysis from the 1H NMR spectrum to be 22 kg mol−1 and 26 kg mol−1 for the Sn(Oct)2 and TBD catalyzed polymerizations respectively. Considering the conversion of L-lactide (98%), the theoretical PLLA arm Mn of the TBD catalyzed polymerization (26 kg mol−1) matches the experimental value, confirming that all PLLA initiated off the macroinitiator. The experimental PLLA arm Mn for the Sn(Oct)2 catalyzed polymerization (91% conversion) is slightly less than the theoretical value (25 kg mol−1). The apparent discrepancy between theory and experiment may be explained by the formation of PLLA homopolymer as evidenced by the low molecular weight peak in the SEC trace (Fig. 5b).
 | ||
| Fig. 6 Representative 1H NMR spectra of (a) CPI-g-HEMI and (b) CPI-g-PLLA. The CPI-g-PLLA contains 95 wt% PLLA grafts off the CPI polymer chain. | ||
Conclusions
We have demonstrated a straightforward catalytic method to produce conjugated dienes post-polymerization on both PI and PCOD that may be implemented on other olefin-containing polymers. The conjugation reaction can be controlled by reaction time and temperature to produce the desired number of conjugated dienes along the backbone of the polymer. Furthermore, these conjugated dienes are reactive towards strong Diels–Alder dienophiles, leading to further functionalized PI and PCOD. Such a “click-chemistry” process can be used to create a multitude of functional groups off the backbone of these conjugated polymers as well as create graft copolymers through a “grafting to” approach. To produce graft copolymers through a “grafting from” approach, CPI was functionalized with hydroxyl groups that served as initiation points for PLLA synthesis. A variety of catalysts polymerized L-lactide off the CPI macroinitiator to produce a potentially tough CPI-g-PLLA graft copolymer.Experimental details
Materials and general methods
All chemicals were purchased from Aldrich and used without any further purification unless otherwise noted. N-2-Hydroxyethyl maleimide was synthesized by a previously published procedure.17L-Lactide (Purac) was purified by recrystallization in ethyl acetate and then dried under vacuum at room temperature. The RuHCl(CO)(PPh3)3 catalyst and P(CH2OH)3 ligand were purchased from Strem Chemicals and used without further purification. HPLC grade toluene and cyclohexanes were dried on a home built solvent column by passing them over an activated alumina column and a supported copper catalyst. HLPC grade CH2Cl2 was dried on an MBraun solvent purification system. All other materials were synthesized as described below.1H NMR spectroscopy was performed on a Varian Inova 500 MHz spectrometer in CDCl3 (Cambridge) using the residual CHCl3 peak as reference. Size exclusion chromatography was performed on an Agilent 1100 high-pressure liquid chromatograph at 35 °C equipped with a PLgel (Varian) 5 μm guard column followed by three PLgel columns with varying pore sizes with HPLC grade chloroform as the mobile phase. Molecular weights and polydispersity index (PDI) were measured by a Hewlett-Packard P1047A refractometer calibrated with polystyrene standards (Polymer Laboratories).
Procedure for anionic polymerization of isoprene
Isoprene was polymerized anionically following a previously published procedure and apparatus setup.43–45 Briefly, isoprene was degassed by three freeze–pump–thaw cycles, dried over n-BuLi twice, and then vacuum distilled to a tared burette. To a flame dried reactor under 5 psig argon atmosphere, the purified isoprene monomer (50 g), dry cyclohexane (800 mL), and sec-BuLi (1.4 M solution in hexanes, 1.28 mL) were added. The reactor was heated at 40 °C in a water bath for 4 h at which time the reaction was quenched with a degassed 50/50 methanol/isopropanol solution. The reaction solution was precipitated in 3× excess 50/50 methanol/isopropanol solution and subsequently dried under vacuum at 45 °C to yield PI (Mn = 25 kg mol−1, 94.2% yield). 1H NMR spectroscopy (500 MHz, CDCl3) δ 5.124 (s, CCH trans and cis-1,4 PI), 4.8–4.6 (m, CCH2 3,4 PI), 2.042 (s, CCH–CH2trans and cis-1,4 PI), 1.678 (s, –CH3cis-1,4 PI), 1.599 (s, –CH3trans-1,4 PI).
General procedure for conjugation of polyisoprene
PI (800 mg) was dissolved in benzene (4.5 mL) in a 20 mL scintillation vial. RuHCl(CO)(PPh3)3 (18 mg) was added to the polymer solution and stirred to create a slurry. The slurry was transferred to a 10 mL side arm pressure vessel. The solution was degassed by three freeze–pump–thaw cycles and backfilled with 3 psig argon. The vessel was then transferred to a 60 °C oil bath to heat for 160 h. Reaction temperatures and times were varied as well as solvents. Toluene and xylenes were substituted for benzene at reaction temperatures higher than 90 °C. At 60 °C under the same reaction conditions, conjugation of PI in either toluene or benzene resulted in similar degrees of conjugation (see ESI†). After the desired reaction time, the solvent was removed by vacuum at ambient temperature over several days. To remove the catalyst, the sealed flask was brought into a N2 atmosphere dry box where dry CH2Cl2 (6.75 mL) and P(CH2OH)3 (23 mg) were added to the flask. The vessel was sealed in the dry box and removed to stir for several days—until the solution had become cloudy white. The cloudy solution was passed through a silica gel column with 150 mL of CH2Cl2. The solution was concentrated by rotary evaporation followed by the addition of BHT (8 mg). The remaining solvent was removed under vacuum at room temperature over several days to give CPI (69.9% yield). See ESI† for complete 1H NMR spectroscopy assignments.General procedure for small molecule and CPI coupling reactions
CPI was dissolved in toluene at 3.3% (w/v) concentration. Subsequently, liquid small molecules were added to the solution by syringe at the desired volume to give the targeted ratio of small molecule to reactive conjugated dienes. For solid small molecules (HEMI and MA), the desired mass was dissolved in minimal CH2Cl2 and then the resulting solution was added to the CPI solution. For reactions to be performed at 160 °C, 5 wt% (relative to CPI) BHT was added to the solution. Once all the components solubilized, the solutions were transferred to side arm pressure vessels where they were degassed by three freeze–pump–thaw cycles and backfilled with 3 psig argon. The sealed reactors were placed in an oil bath at the desired temperature (110 °C or 160 °C) to react for varying times. Upon completion, the flasks were removed from the oil bath to cool to ambient temperature. The resulting solutions were precipitated three times from CH2Cl2 into 10× excess methanol. The product was dissolved in CH2Cl2 and 1 wt% BHT was added to the solution. The solution was concentrated with blowing N2 and dried under vacuum at room temperature for several days. Products were analyzed with 1H NMR spectroscopy and SEC. See ESI† for detailed 1H NMR spectra assignments and SEC traces.Procedures for CPI-g-PLLA synthesis
CPI-g-HEMI with 19 hydroxyl groups per molecule was synthesized using the above method for Diels–Alder reactions with CPI and subsequently used as the macroinitiator for CPI-g-PLLA synthesis. All reactions targeted 95 wt% L-lactide monomer (0.475 g) and 5 wt% macroinitiator (25 mg) were setup in a N2 dry box, and were performed in 48 mL pressure vessels. After the polymerization reaction, solutions were diluted in CH2Cl2 and precipitated in 10× excess methanol. The precipitated polymers were collected by suction filtration and dried under vacuum at room temperature until dry. The specific synthetic details for each catalyst used are as follows. Sn(Oct)2catalyzed reaction. The macroinitiator and monomer were dissolved in dry toluene (3.5 mL). Sn(Oct)2 was added at a monomer to catalyst ratio of 5000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as a stock solution of Sn(Oct)2 in toluene. The reaction vessel was heated at 100 °C for 21 h in an oil bath, after which the vessel was removed from heat and allowed to cool to ambient temperature to quench the reaction. AlEt3catalyzed reaction. The macroinitiator was dissolved in dry toluene (4.1 mL). AlEt3 solution (1 M in hexanes) was added at a 2:1 hydroxyl group to catalyst ratio (7.8 μL) and the resulting solution was left to stir overnight in the dry box for 16 h. Subsequently, the monomer was added to the solution and the vessel was placed in a 90 °C oil bath for 5 h. One drop of acid water (4:1 DI water to concentrated HCl) was added to quench the reaction. TBD catalyzed reaction. The macroinitiator and monomer were dissolved in dry CH2Cl2. TBD (0.5 mg) was added as a stock solution in dry CH2Cl2. The solution was allowed to react for 4 h at room temperature and then quenched with benzoic acid (4.3 mg) solution in CH2Cl2. PLLA repeat unit 1H NMR spectroscopy assignments (500 MHz, CDCl3) δ 5.158 (q, J = 7.2 Hz, O–CH–CH3), 1.576 (d, J = 8.1 Hz, O–CH–CH3).
:1 as a stock solution of Sn(Oct)2 in toluene. The reaction vessel was heated at 100 °C for 21 h in an oil bath, after which the vessel was removed from heat and allowed to cool to ambient temperature to quench the reaction. AlEt3catalyzed reaction. The macroinitiator was dissolved in dry toluene (4.1 mL). AlEt3 solution (1 M in hexanes) was added at a 2:1 hydroxyl group to catalyst ratio (7.8 μL) and the resulting solution was left to stir overnight in the dry box for 16 h. Subsequently, the monomer was added to the solution and the vessel was placed in a 90 °C oil bath for 5 h. One drop of acid water (4:1 DI water to concentrated HCl) was added to quench the reaction. TBD catalyzed reaction. The macroinitiator and monomer were dissolved in dry CH2Cl2. TBD (0.5 mg) was added as a stock solution in dry CH2Cl2. The solution was allowed to react for 4 h at room temperature and then quenched with benzoic acid (4.3 mg) solution in CH2Cl2. PLLA repeat unit 1H NMR spectroscopy assignments (500 MHz, CDCl3) δ 5.158 (q, J = 7.2 Hz, O–CH–CH3), 1.576 (d, J = 8.1 Hz, O–CH–CH3).
Acknowledgements
The authors would like to acknowledge Louis M. Pitet for the synthesis of polycyclooctadiene polymer and support from the Center for Sustainable Polymers at the University of Minnesota—funded by a grant from the Initiative for Renewable Energy and the Environment at the University of Minnesota.References
- G. M. Whited, F. J. Feher, D. A. Benko, M. A. Cervin, G. K. Chotani, J. C. McAuliffe, R. J. LaDuca, E. A. Ben-Shoshan and K. J. Sanford, Ind. Biotechnol., 2010, 6, 152–163 Search PubMed.
- M. P. McGrath, E. D. Sall and S. J. Tremont, Chem. Rev., 1995, 95, 381–396 CrossRef CAS.
- D. Derouet, Q. N. Tran and H. H. Thuc, Eur. Polym. J., 2007, 43, 1806–1824 Search PubMed.
- M. L. Hallensleben, H. Rüdiger Schmidt and R. H. Schuster, Angew. Makromol. Chem., 1995, 227, 87–99 Search PubMed.
- D. Derouet, F. Morvan and J.-C. Brosse, Eur. Polym. J., 2001, 37, 1297–1313 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demiral, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83–98 Search PubMed.
- S. Sinnwell, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Block, Stars, and Combs: Complex Macromolecular Architecture Polymers via Click Chemistry, in Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley and Sons, Ltd, UK, 2009, pp. 89–117 Search PubMed.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191–198 CrossRef CAS.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- W. M. Gramlich, M. L. Robertson and M. A. Hillmyer, Macromolecules, 2010, 43, 2313–2321 Search PubMed.
- A. Dag, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 46, 302–313 Search PubMed.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499–506 CrossRef CAS.
- L. Dai and J. W. White, Polymer, 1991, 32, 2120–2127 CAS.
- M. Thakur, Macromolecules, 1988, 21, 661–664 CrossRef CAS.
- E. D. Owen and H. S. M. Al-Moh'd, Polymer, 1997, 38, 3533–3538 Search PubMed.
- L. Dai, A. W. H. Mau, H. J. Griesser and D. A. Winkler, Macromolecules, 1994, 27, 6728–6735 CrossRef CAS.
- M. Thakur, R. Swamy and J. Titus, Macromolecules, 2004, 37, 2677–2678 Search PubMed.
- L. Dai and A. W. H. Mau, Synth. Met., 1997, 86, 1893–1894 Search PubMed.
- G.-Q. Yu and M. Thakur, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 2099–2104 Search PubMed.
- J. K. McLean, S. A. Guillen-Castellanos, J. S. Parent, R. A. Whitney and R. Resendes, Eur. Polym. J., 2007, 43, 4619–4627 Search PubMed.
- S. M. Malmberg, J. S. Parent, D. A. Pratt and R. A. Whitney, Macromolecules, 2010, 43, 8456–8461 Search PubMed.
- N. Kuźnik and S. Krompiec, Coord. Chem. Rev., 2007, 251, 222–233 CrossRef CAS.
- B. Schmidt, Eur. J. Org. Chem., 2004, 2004, 1865–1880 Search PubMed.
- N. Kuźnik, S. Krompiec, T. Bieg, S. Baj, K. Skutil and A. Chrobok, J. Organomet. Chem., 2003, 665, 167–175 CrossRef CAS.
- S. Krompiec, M. Krompiec, R. Penczek and H. Ignasiak, Coord. Chem. Rev., 2008, 252, 1819–1841 CrossRef CAS.
- B. Alcaide, P. Almendros and A. Luna, Chem. Rev., 2009, 109, 3817–3858 CrossRef CAS.
- S. Krompiec, N. Kuźnik, M. Krompiec, R. Penczek, J. Mrzigod and A. Tórz, J. Mol. Catal. A: Chem., 2006, 253, 132–146 CrossRef CAS.
- D. Gauthier, A. T. Lindhardt, E. P. K. Olsen, J. Overgaard and T. Skrydstrup, J. Am. Chem. Soc., 2010, 132, 7998–8009 CrossRef CAS.
- S. Krompiec, R. Penczek, M. Krompiec, T. Pluta, H. Ignasiak, A. Kita, S. Michalik, M. Matlengiewicz and M. Filapek, Curr. Org. Chem., 2009, 13, 896–913 Search PubMed.
- R. C. Larock, X. Dong, S. Chung, C. K. Reddy and L. E. Ehlers, J. Am. Oil Chem. Soc., 2001, 78, 447–453 CrossRef CAS.
- D. D. Andjelkovic, B. Min, D. Ahn and R. C. Larock, J. Agric. Food Chem., 2006, 54, 9535–9543 Search PubMed.
- G. Theryo, F. Jing, L. M. Pitet and M. A. Hillmyer, Macromolecules, 2010, 43, 7394–7397 Search PubMed.
- H. Wakamatsu, M. Nishida, N. Adachi and M. Mori, J. Org. Chem., 2000, 65, 3966–3970 CrossRef CAS.
- C. J. Yue, Y. Liu and R. He, J. Mol. Catal. A: Chem., 2006, 259, 17–23 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, John Wiley and Sons, New York, 5th edn, 2001, pp. 1062–1075 Search PubMed.
- S. Ndoni, C. M. Papadakis, F. S. Bates and K. Almdal, Rev. Sci. Instrum., 1995, 66, 1090–1095 CrossRef CAS.
- M. A. Hillmyer and F. S. Bates, Macromolecules, 1996, 29, 6994–7002 CrossRef CAS.
- S. C. Schmidt and M. A. Hillmyer, Macromolecules, 1999, 32, 4794–4801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed 1H NMR spectra and peak assignments for PI, CPI, small molecule grafts to CPI, and PI-g-PLLA. Also described, synthesis and characterization of CPCOD, small molecule grafts to CPCOD, and HEMI-PLLA grafts to CPI. See DOI: 10.1039/c1py00147g |
| This journal is © The Royal Society of Chemistry 2011 |