DOI:
10.1039/C1PY00140J
(Paper)
Polym. Chem., 2011,
2, 1986-1991
Efficient dual radical/cationic photoinitiator under visible light: a new concept
Received
30th March 2011
, Accepted 31st May 2011
First published on 13th June 2011
Abstract
A new concept based on the photoredox catalysis is proposed for the design of a photoinitiating system able to efficiently generate, in a single catalytic cycle, both a radical and a cation as initiating species. This is exemplified here by the tris(1,10-phenanthroline)ruthenium(II) (Ru(phen)32+) complex in combination with a diphenyl iodonium salt and a silane. Interesting and promising photopolymerization profiles in free radical photopolymerization (FRP) and free radical promoted cationic photopolymerization (FRPCP) are obtained under air and upon xenon lamp exposure and even soft irradiation conditions (fluorescence bulb, sunlight). An acrylate/epoxide blend is also easily polymerized under air using a fluorescent bulb. The mechanisms are investigated by ESR and Laser Flash Photolysis experiments.
Introduction
With the existing demand for green technologies, the development of new photoinitiating systems (PIS) is required.1 Besides the usual basic properties (reactivity, efficiency, solubility, compatibility, lack of toxicity…), PISs must also exhibit challenging specific properties, for example i) a good radical initiating ability under visible light and especially soft irradiation conditions (sunlight, ambient light, fluorescence bulbs…) under air, ii) catalytic behavior of the photoinitiator (PI) allowing its use in low quantities (this approach is also related to the “economy of atoms” concept), iii) access to a large range of photopolymerizable formulations (including natural and renewable monomers) under mild and convenient synthetic conditions, iv) an ability to work either separately in free radical photopolymerization (FRP) and free radical promoted cationic photopolymerization (FRPCP) or simultaneously in FRP/FRPCP.1
Free radical polymerization and cationic polymerization are usually achieved (see e.g.ref. 2 and references therein) by two different PIs (a radical PI and a cationic PI). This drawback becomes obviously more important for visible light: cationic PIs usually absorb in the UV. Therefore, the light source must contain a reasonable amount of UV light (such as in Hg, Xe, Hg doped lamps). At present, such photoinitiating systems exhibiting almost similar properties for both radical and cationic processes (for λ > 400 nm) remain highly desirable e.g. under sun or fluorescent bulbs.
The results obtained in our very recent work3 where Ru(bpy)32+ (tris(2,2′-bipyridine)ruthenium(II) dichloride hexahydrate) was presented as an interesting PI, mostly for FRPCP, under soft and visible light irradiations under air prompted us to propose here a new concept, summarized in Scheme 1, that could meet the challenge detailed above. The mechanism corresponds to photoredox catalysis. The first step corresponds to the excitation of a suitable starting PI (sPI) by a visible light which then reacts with a radical source to generate an initiating radical R˙ for FRP and a transient PI (tPI). In a second step, an interaction between R˙ and tPI leads to a cation C+ and regenerates the starting PI ground state sPI (tPI must be a good oxidation agent). If R˙ and C+ are efficiently and rapidly formed under a Xe lamp, sun or a fluorescent bulb, the system should be operative for FRP using an acrylate or FRPCP using an epoxide.
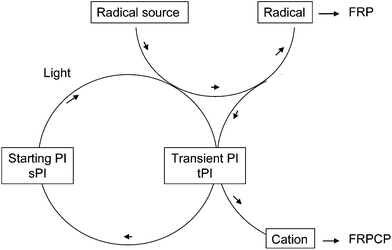 |
| | Scheme 1 | |
In the present paper, such a dual photosensitive system will be exemplified by Ru(phen)32+ (where phen stands for a phenanthroline ligand) as the sPI, a silane and an iodonium salt as the radical sources. Due to its properties (light absorption and rather bad solubility in acrylate matrix), the Ru(bpy)32+ complex proposed in ref. 3 or introduced recently in organic photocatalyzed synthesis4–10 cannot be really a candidate here as an efficient dual PI. Selected examples for the FRP of acrylates, the ring opening polymerization of epoxides by FRPCP and the simultaneous FRP/FRPCP of an acrylate/epoxide blend will be provided and the involved mechanisms investigated by Laser Flash Photolysis and ESR spin trapping experiments. A comparison of Ru(phen)32+ with other complexes (Ru(bpy)32+ or Fe(phen)32+) will illustrate the key role of sPI to act as a true dual photoinitiator.
Experimental part
i) Compounds
Tris(1,10-phenanthroline)ruthenium(II) (Ru(phen)32+), tris(1,10-Phenanthroline)iron(II) perchlorate (Fe(phen)32+), tris(trimethylsilyl)silane (TTMSS) and diphenyl iodonium hexafluorophosphate (Ph2I+) were obtained from Aldrich and used at the best purity available. The acrylate monomers, trimethylolpropane triacrylate (TMPTA) and ethoxylated pentaerythritol tetraacrylate (EPT) from Cray Valley, were selected. For the ring opening polymerization (3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX from Cytec; Uvacure 1500), limonene dioxide (LDO) from Millennium Speciality Chemicals and epoxidized soybean oil (ESO) from Arkema (Ecepox; epoxy content: 3.7 M kg−1) were chosen as epoxy monomers.
ii) Free radical photopolymerization (FRP) experiments
For film polymerization experiments, TMPTA or EPT were used as low viscosity monomers. The experiments were carried out in laminate and under air conditions. The films (20 μm thick) deposited on a BaF2 pellet were irradiated (see figure captions). The evolution of the double bond content was continuously followed by real time FTIR spectroscopy (Nexus 870, Nicolet) at about 1630 cm−1.11
The two- and three-component photoinitiating systems are based (except where otherwise stated) on Ru(phen)32+/Ph2I+ (0.2%/2% w/w) and Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w). The epoxide films (25 μm thick) deposited on a BaF2 pellet were irradiated under air inside the IR spectrometer cavity. The evolution of the epoxy group content at about 790 cm−1 is continuously followed by real time FTIR spectroscopy (see above).
iv)
ESR spin trapping (ESR-ST) experiments
ESR-ST experiments were carried out using a X-Band spectrometer (MS 200 Magnettech). The radicals were produced at RT under a Xenon lamp exposure (except otherwise noted) and trapped by phenyl-N-tbutylnitrone (PBN) according to a procedure described in detail in ref. 12.
The nanosecond laser flash photolysis LFP experiments were carried out with a Q-switched nanosecond Nd/YAG laser at λexc = 355 nm (9 ns pulses; energy reduced down to 10 mJ; Powerlite 9010 Continuum), the analyzing system consisted of a pulsed xenon lamp, a monochromator, a fast photomultiplier and a transient digitizer.12a
Results and discussion
A) Photochemical properties
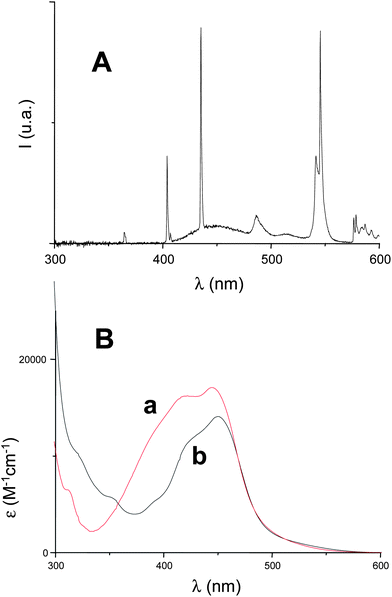
Interestingly, the Ru(phen)32+ complex exhibits better light absorption properties than Ru(bpy)32+ in the 350–600 nm range (Fig. 1). The excitation of Ru(phen)32+ results in the formation (1) of a known13 relatively long lived emissive excited state that can be observed at about 620 nm (Fig. 2A). This state is efficiently quenched (Fig. 2A) by the iodonium salt Ph2I+ (k = 4.9 × 107 M−1s−1) but very slowly deactivated by TTMSS (k < 5 × 105 M−1s−1). The free-energy change (ΔGEt) for the electron transfer from a donor to an acceptor can be calculated from the classical Rehm-Weller equation.14 Therefore, reaction (2) is favorable according to the redox properties of these reactants (Eox(Ru(phen)32+) = 1.3 V; Ered(Ph2I+) ≈ −0.2 V; E(*Ru(phen)32+) = 2.15 eV; free energy change ΔG = −0.65 eV).13,14| | | Ru(phen)32+ → → → *Ru(phen)32+ (hν) | (1) |
| | | *Ru(phen)32+ + Ph2I+ → Ru(phen)33+ + Ph˙ + Ph–I | (2) |
| | | Ph˙ + (TMS)3Si–H → Ph–H + (TMS)3Si˙ | (3) |
| | | R3Si˙ + Ph2I+ → R3Si+ + Ph˙ + Ph–I | (4a) |
| | | R3Si˙ + Ru(phen)33+ → Ru(phen)32+ + R3Si+ | (4b) |
Process (2) is also well evidenced here through the observation of Ph˙i.e. in ESR-spin trapping experiments (Fig. 2B), the observed hyperfine coupling (hfc) constants (aN = 14.5 G; aH = 2.45 G) being in full agreement with the known data for this radical.15Phenyl radicals can also be generated in (4a): the oxidation rate constant of tris(trimethylsilyl)silyl by Ph2I+ was already determined (2.6 × 106 M−1s−1 in ref. 11a).
![(A) Luminescence decay (recorded at 620 nm) for Ru(phen)32+ in acetonitrile for different [Ph2I+]. (B)ESR spectrum obtained after light irradiation of Ru(phen)32+/Ph2I+. In tert-butyl benzene/acetonitrile (50/50); PBN is used as spin-trap; [Ph2I+] = 0.011 M under argon.](/image/article/2011/PY/c1py00140j/c1py00140j-f2.gif) |
| | Fig. 2
(A) Luminescence decay (recorded at 620 nm) for Ru(phen)32+ in acetonitrile for different [Ph2I+]. (B)ESR spectrum obtained after light irradiation of Ru(phen)32+/Ph2I+. In tert-butyl benzene/acetonitrile (50/50); PBN is used as spin-trap; [Ph2I+] = 0.011 M under argon. | |
Upon irradiation of Ru(phen)32+/TTMSS/Ph2I+, the formation of silyl radicals (characterized by the hfc aN = 15.0 G and aH = 5.9 G in agreement with ref. 15) is well observed in ESR-ST experiments. This is fully consistent with the formation of silyl radicals (R3Si˙) from a hydrogen abstraction reaction (3) on TTMSS by the Ph˙ radical generated in (2). This process is also shown by the Si–H conversion at about 2050 cm−1 in polymerization experiments.
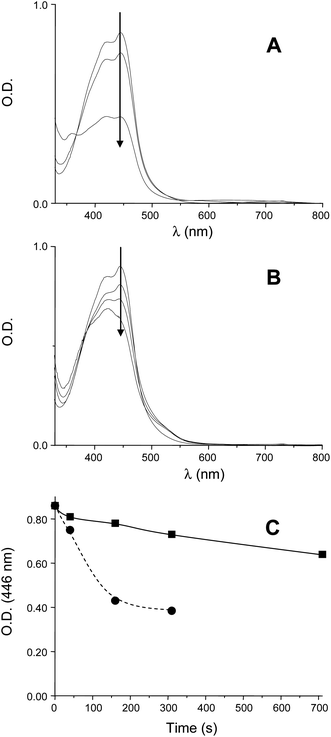
A slower photolysis (Fig. 3) is observed in Ru(phen)32+/TTMSS/Ph2I+ compared to Ru(phen)32+/Ph2I+: Ru(phen)32+ behaves like a photocatalyst and it is largely recovered in the presence of TTMSS during the light exposure. The oxidation of the silyl radicals R3Si˙ by Ru(phen)33+(4b) regenerating the starting compound (Ru(phen)32+) likely accounts for this behavior. This also ensures that (4b) is probably the major process for silyl radical oxidation.
 |
| | Fig. 3
(A) Photolysis of Ru(phen)32+/Ph2I+ in acetonitrile (4.7 × 10−5 M/2.3 × 10−3 M); UV-visible spectra for different irradiation times: t = 0 s; t = 40 s and t = 160 s. (B) Photolysis of Ru(phen)32+/TTMSS/Ph2I+ in acetonitrile (4.7 × 10−5 M/4.5 × 10−2 M/2.3 × 10−3 M); UV-visible spectra for different irradiation times: t = 0 s; t = 40 s; t = 310 s and 710 s. (C) O.D. at 446 nm vs. time for (A): circles and (B): squares. | |
As summarized in reactions (1–4), the proposed system can generate both radical (Ph˙, R3Si˙) and cationic (R3Si+) species: these routes can be affected by the competitive reaction pathways involving the monomer.
The best conversion-time profiles and the best systems suitable for different irradiation sources are shown as examples in Fig. 4–6.
Free radical polymerization (FRP).
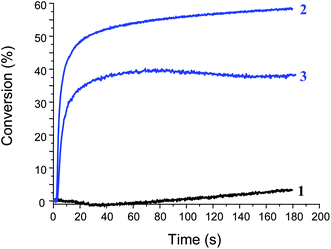
Interestingly, Ru(phen)32+/TTMSS/Ph2I+ is an excellent initiating system i.e. high polymerization rates and final conversions of TMPTA and EPT are obtained both in laminate and under air (Fig. 4) under a Xe lamp. Using bis (η6 5–2,4-cyclopentadien-1-yl) bis[2,6-difluoro-3-(1H-pyrrol-1-yl) phenyl]titanium (or Irgacure 784), a well known type I photoinitiator under visible lights, only a low final conversion (<10%) is noted under air upon a Xenon lamp irradiation. On the opposite, a conversion of about 40% is reached here using the Ru(phen)32+/TTMSS/Ph2I+ system (Fig. 4) thereby demonstrating the high reactivity of the new proposed PIS. This is in line with the excellent polymerization initiation ability of the generated radicals: indeed, Ph˙ and (TMS)3Si˙ exhibit high addition rate constants to acrylate double bonds (k > 106 M−1s−1).16 The ability of TTMSS to overcome the oxygen inhibition in FPR, as described in ref. 17, explains the high efficiency obtained in aerated conditions. A similar behavior is still found upon laser diode (532 nm) irradiation. Upon fluorescent bulb irradiation under air, polymerization is hard (conversion < 15%): this can be ascribed to the low light intensity of this specific irradiation device.3
 |
| | Fig. 4
Polymerization profiles of TMPTA upon a Xenon lamp irradiation (λ > 390 nm) in the presence of (1) Ru(phen)32+ (0.2% w/w) in laminate; (2) Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w) in laminate; (3) Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w) under air. | |
Cationic polymerization (CP).
The low polymerization ability of the Ru(phen)32+/Ph2I+ system highlights the lack of efficient cationic initiating structure in the absence of silane. The addition of TTMSS drastically improves the polymerization profiles under a Xe lamp exposure under air (Fig. 5–6). This is ascribed to the formation of silylium cations (R3Si+) which are very efficient towards the ring-opening process of epoxides:11
Interestingly, under a fluorescent bulb irradiation, the Ru(phen)32+/TTMSS/Ph2I+ system is efficient (Fig. 5; the formed polyether network is easily characterized by its absorption band at 1080 cm−1). An almost complete conversion even for these low light energy irradiations is reached in 10 min. A similar behavior is still found for the Xenon lamp, the laser diode (532 nm) irradiations (Fig. 6) and also for sunlight (Mulhouse; France - October 2010). Interestingly, for this initiating system (Ru(phen)32+/TTMSS/Ph2I+), the polymerization of renewable epoxy monomers can be performed (epoxidized soybean oil and limonene dioxide) under this fluorescence bulb irradiation (conversion of 40% and 65% for 20 min of irradiation for ESO and LDO, respectively). This is highly worthwhile for green chemistry applications i.e. the use of renewable monomers can be combined with low energy requirements (fluorescence bulbs, sunlight). The same process based on Ru(bpy)32+ was harder (see below).
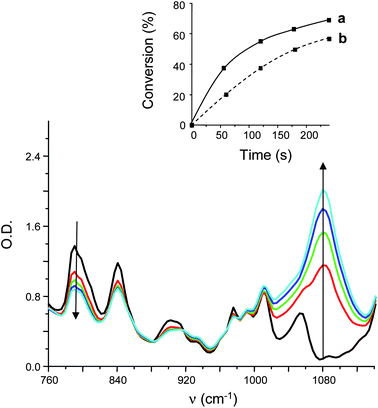 |
| | Fig. 5
IR spectra recorded during the photopolymerization of EPOX in the presence of Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w) under fluorescence bulb irradiation for different irradiation times (t = 0 to t = 3 min). Insert: monomer conversion vs. time profile (under air) for (a) Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w) and (b) Ru(bpy)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w). | |
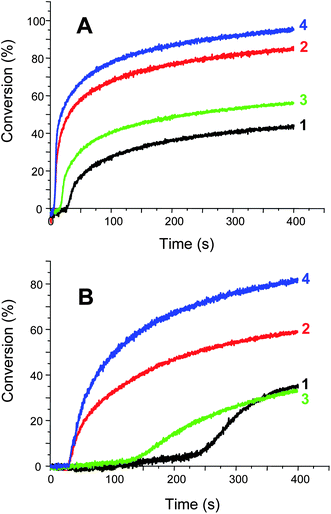 |
| | Fig. 6
Photopolymerization profiles of EPOX. (A) upon Xenon lamp irradiation (λ > 390 nm) in the presence of (1) Ru(bpy)32+/Ph2I+ (0.2%/2% w/w); (2) Ru(bpy)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w); (3) Ru(phen)32+/Ph2I+ (0.2%/2% w/w); (4) Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w). (B) upon a diode laser irradiation (532 nm) in the presence of (1) Ru(bpy)32+/Ph2I+ (0.2%/2% w/w); (2) Ru(bpy)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w); (3) Ru(phen)32+/Ph2I+ (0.2%/2% w/w); (4) Ru(phen)32+/TTMSS/Ph2I+ (0.2%/3%/2% w/w). | |
Recently, other initiating systems have been proposed for visible lights (see ref. 19 and references therein); the systems proposed here exhibit quite good reactivity.
Hybrid radical and cationic photopolymerization.
The proposed system is able to initiate both radical and cationic polymerization processes, so its ability to polymerize a mixture of TMPTA and EPOX (50%/50% w/w) in a one-step hybrid cure process upon an exposure to a fluorescence bulb and under air was investigated (Fig. 7). Interestingly, a tack free coating is obtained for only 3.5 min of fluorescence bulb irradiation under air. At this time, the acrylate and epoxide conversions are ∼65% and ∼45% (from their respective IR bands). The characterization of the network is beyond the scope of the present paper.
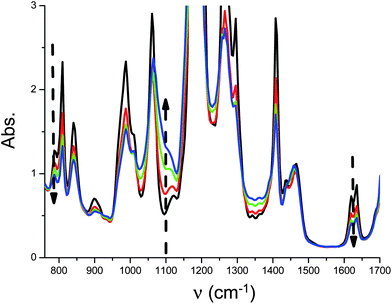 |
| | Fig. 7
IR spectra recorded during the photopolymerization of (3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate/TMPTA (50%/50% w/w) under air in the presence of Ru(phen)32+/TTMSS/Ph2I+ (0.1%/3%/2% w/w); fluorescence bulb irradiation at different times (t = 0 s; 10 s; 40 s and 3 min 30 s). The evolution of the acrylate double bond, epoxide function and polyether group are observed at about 1630 cm−1, 790 and 1080 cm−1, respectively. | |
Role of other photoinitiators.
For sake of comparison, other photoinitiating systems were checked. Fe(phen)32+ is characterized by an intense absorption at about 508 nm (ε > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1cm−1).18Fe(phen)32+/TTMSS/Ph2I+ is found not to be a very efficient initiating system for FRP or CP processes with final monomer conversions under air of <10% and 25% for TMPTA and EPOX, respectively (irradiation 400 s; Xenon lamp; λ > 400 nm). This can be partly ascribed to the shorter excited state lifetime for this iron derivative (∼800 ps)18 compared to Ru(phen)32+ (∼450 ns) which does not allow an efficient iodonium salt reduction and the associated Ph˙ formation (reaction 2).
000 M−1cm−1).18Fe(phen)32+/TTMSS/Ph2I+ is found not to be a very efficient initiating system for FRP or CP processes with final monomer conversions under air of <10% and 25% for TMPTA and EPOX, respectively (irradiation 400 s; Xenon lamp; λ > 400 nm). This can be partly ascribed to the shorter excited state lifetime for this iron derivative (∼800 ps)18 compared to Ru(phen)32+ (∼450 ns) which does not allow an efficient iodonium salt reduction and the associated Ph˙ formation (reaction 2).
The molar extinction coefficients for the maximal absorption wavelength are noticeably higher i.e. about 18000 M−1cm−1 for Ru(phen)32+vs. 14 500 M−1 cm−1 for Ru(bpy)32+ (Fig. 1B in acetonitrile). From the comparison with the emission spectrum of the fluorescence bulb (Fig. 1A), the absorption of the two intense bands at 405 and 435 nm will be much better for the phenanthroline derivative. In the 300–700 nm wavelength range, the light absorbed intensity (Iabs) was calculated according to eqn (1) where I0 and OD (which depend on the wavelength) stand for the light source intensity I0 at the sample (obtained by an absolute irradiance measurement with the Ocean Optics HR4000) and the absorbance of the ruthenium complexes (OD).
| | | Iabs = ∫I0(1 − 10−OD) dλ | (eqn 1) |
Through eqn (1), the light absorption is 20% higher for Ru(phen)32+ compared to Ru(bpy)32+: this can be highly worthwhile for the use of Ru(phen)32+ as a photocatalyst for these soft irradiation conditions.
The polymerization profiles of TMPTA or EPT as well as those of EPOX are better than those obtained with the Ru(bpy)32+ complex in ref. 3 (see also Fig. 5–6). This is ascribed both to the better light absorption properties of Ru(phen)32+ and also its better solubility in acrylates.
The redox and excited state properties of Ru(phen)32+ and Ru(bpy)32+ are similar.13 The free-energy change (ΔGEt) is found to be favorable for reaction (2) and the rate constants are >107–108 M−1s−1. The better light absorption properties of the phenanthroline derivative probably ensures that this compound is more appropriated than Ru(bpy)32+ for its use as a photocatalyst under soft irradiation. Its application in organic synthesis can also be worthwhile i.e. only the bpy derivative is actually used.4–10
Conclusions
In the present paper, a Ru(phen)32+ complex is proposed as a photoinitiator for the initiation of free radical or cationic polymerization processes under air and upon irradiation with a visible light xenon lamp, the sun or a household green bulb. Ru(phen)32+ behaves like a photocatalyst as it is recovered during light exposure. The present Ru(phen)32+/silane/iodonium salt combination is better than a titanocene based system in FRP11c and other previously proposed multi-component systems in FRPCP.11c It also allows an efficient hybrid cure of an acrylate/epoxide formulation upon a green fluorescence bulb irradiation (both monomers exhibit a high conversion at the same exposure time). In our opinion, this paper opens a way to design efficient dual photoinitiating systems by selecting other PI and radical sources: new photocatalysts based on different metals will be also proposed.
Acknowledgements
This work was supported by the “Agence Nationale de la Recherche” ANR under Grant ANR-10-BLAN-0802 (SILICIUM 2010).
References
-
(a)
J. P. Fouassier, Photoinitiation, Photopolymerization and Photocuring: Fundamental and Applications, Hanser Publishers, New-York, 1995 Search PubMed;
(b)
Photoinitiated Polymerization, ed. K. D. Belfield, J. V. Crivello, ASC Symposium series 847, 2003 Search PubMed;
(c)
Photochemistry and UV Curing, ed. J. P. Fouassier, Researchsignpost, Trivandrum India, 2006 Search PubMed.
-
(a) M. Sangermano, W. Carbonaro, G. Mallucelli and A. Priola, Macromol. Mater. Eng., 2008, 293, 515–520 Search PubMed;
(b) L. Lecamp, C. Pavillon, P. Lebaudy and C. Bunuel, Eur. Polym. J., 2005, 41, 169–175 Search PubMed;
(c) C. Rajaraman, W. A. Mowers and J. V. Crivello, Macromolecules, 1999, 32, 36–41 Search PubMed;
(d) C. Decker and T. Bendaikha, J. Appl. Polym. Sci., 1998, 70, 2269–2282 CrossRef CAS;
(e) C. Decker, T. Nguyen Thi Viet and H. Le Xuan, Eur. Polym. J., 1996, 32, 1319–1326 CrossRef CAS.
- J. Lalevée, N. Blanchard, M.-A. Tehfe and J. P. Fouassier, Macromolecules, 2010, 43, 10191–10195 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS.
- J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS.
- L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS.
- J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985–4987 RSC.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS.
-
(a) J. Lalevée, M. El-Roz, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2008–2014 CrossRef CAS;
(b) J. Lalevée, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3042–3047 Search PubMed;
(c) M.-A. Tehfe, J. Lalevée, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8669–8674 CrossRef;
(d) M.-A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS;
(e) M.-A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1830–1837 CrossRef CAS.
-
(a) J. Lalevée, N. Blanchard, M. El-Roz, B. Graff, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 4180–4186 CrossRef CAS;
(b) D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef CAS.
- A. Kapturkiewicz, P. Szrebowaty, G. Angulo and G. Grampp, J. Phys. Chem. A, 2002, 106, 1678–1685 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CAS.
-
(a)
Landolt Bornstein: Magnetic Properties of Free Radicals, ed. H. Fischer, Springer Verlag, Berlin, 2005, vol. 26d Search PubMed;
(b) H. Chandra, I. M. T. Davidson and M. C. R. Symons, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2705–2711 RSC.
-
(a)
C. Chatgilialoglu, Organosilanes in Radical Chemistry, John Wiley & Sons, Chichester, 2004 Search PubMed;
(b) J. Lalevée, X. Allonas and J. P. Fouassier, J. Phys. Chem. A, 2004, 108, 4326–4334 CrossRef CAS.
- M. El-Roz, J. Lalevée, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS.
-
(a) A. J. Street, D. M. Goodall and R.-C. Greenhow, Chem. Phys. Lett., 1978, 56, 326–329 CrossRef CAS;
(b) C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309–1319 CrossRef CAS.
-
(a) J. V. Crivello and M. Sangermano, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 343–356 CrossRef CAS;
(b) J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl. Chem., 2009, 46, 474–483 Search PubMed;
(c) J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866–875 CrossRef CAS;
(d) J. V. Crivello and U. Bulut, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5217–5321 CrossRef CAS;
(e) M. Degirmenci, A. Onen, Y. Yagci and S. P. Pappas, Polym. Bull., 2001, 46, 443–449 CrossRef CAS;
(f) Y. Y. Durmaz, N. Moszner and Y. Yagci, Macromolecules, 2008, 41, 6714–6718 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.