Simple and efficient copper metal-mediated synthesis of alkoxyamine initiators†
Simon
Harrisson
,
Patrick
Couvreur
and
Julien
Nicolas
*
Laboratoire de Physico-Chimie, Pharmacotechnie et Biopharmacie, Univ Paris-Sud, UMR CNRS 8612, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296, Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Fax: +33 1 46 83 59 46; Tel: +33 1 46 83 58 53
First published on 27th May 2011
Abstract
A simple and efficient method is presented for the preparation of a wide range of alkoxyamines from nitroxide radicals and activated alkyl bromides at room temperature. The reaction requires a stoichiometric amount of copper metal (0.5 mol mol−1 alkyl bromide) and proceeds most rapidly in polar solvents such as acetonitrile, dimethyl sulfoxide or ethanol. No addition of copper(I) or copper(II) salts is necessary. Isolated yields of 71–94% were obtained across a range of alkyl bromide and nitroxide substrates, with reaction times of 1–16 h. The method is compatible with carboxylic acid and amine functionalities.
Introduction
Alkoxyamines of the form R1–ONR2R3, where R1 is a good homolytic leaving group, undergo reversible thermal homolysis of the R1–O bond to produce reactive R1˙ and persistent R2R3NO˙ free radicals. These compounds have found several applications in organic chemistry, including in the formation of heterobivalent libraries,1 radical cyclization2 and intermolecular addition reactions.2 The major application of alkoxyamines, however, is in nitroxide-mediated polymerization (NMP), a leading technique for the control of radical polymerizations.3 Early drawbacks of NMP, such as the requirement for high temperatures and applicability to a limited number of monomers, have been substantially overcome by the development of more active alkoxyamines derived from 2,2,5-trimethyl-4-phenyl-3-azahexane-3-oxyl (TIPNO)4 or N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide (SG1)5 (Scheme 1).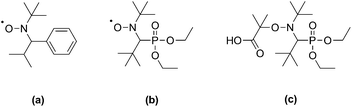 | ||
| Scheme 1 Structures of TIPNO (a) and SG1 (b) nitroxides, and of the SG1-based BlocBuilder MA alkoxyamine (c). | ||
These second-generation alkoxyamines have been successfully used to control the polymerization of styrenic and acrylic monomers with good control over molecular weight. In particular, N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl) hydroxylamine (BlocBuilder MA, Scheme 1), based on SG1, is one of the most potent alkoxyamines developed so far. Its use has led to significant advances in the control of bulk/solution6–8 and emulsion9 polymerizations, and the preparation of functionalized polymers.10–13
Several routes have been developed for the synthesis of alkoxyamines,14,15 of which the most prominent are nucleophilic substitution of a hydroxylaminate anion on an alkyl halide, or the combination of an alkyl radical (generated in situ) with a stable nitroxide radical. The first method requires the preparation of a hydroxylamine and its anion. These compounds are generally quite unstable, and so this pathway has seen limited use, restricted to the preparation of some 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) and di-tert-butyl nitroxide derivatives.
The second route is perhaps the most convenient and popular method for preparing alkoxyamines. The alkyl radical may be generated by thermolysis16 or photolysis17 of a radical initiator, or by homolytic scission of alkyl halides. The latter method was developed by Matyjaszewski et al.,18 who applied a concept from atom transfer radical polymerization (ATRP)19,20 to generate alkyl radicals by reaction of an activated alkyl bromide with CuBr (generated in situ by comproportionation of Cu metal and CuBr2). In this approach, the radicals were trapped by TEMPO or a related nitroxide to form the desired alkoxyamine. The versatility, high yield, simple purification and ready availability of brominated substrates have resulted in this method or variations thereof becoming one of the most popular for the production of alkoxyamines.1,21–30 A related technique involved the use of CuBr to generate polymeric radicals from ω-bromo polymers which were trapped in situ by functionalized nitroxides with high efficiency.31–36 However, the method is not without drawbacks: the original procedure18 was performed at elevated temperatures (55–75 °C) over 4–18 h, which would result in the decomposition of more labile SG1-based alkoxyamines. An optimized procedure21–23 gave good yields at room temperature but required the use of a 3-fold excess of copper (1–1.5 eq. Cu, 1.5–2 eq. CuBr relative to nitroxide) and excess activated alkyl bromide (1.5–2 eq. relative to nitroxide).
Recently, Percec et al. reported the controlled polymerization of acrylates and methacrylates mediated by Cu metal in dimethyl sulfoxide (DMSO).37 The polymerizations were rapid, with near complete conversion obtained in 10–30 minutes at room temperature. This system has since been extended to other solvents38,39 and monomers.40–42 These results, as well as some earlier reports of the use of Cu/alkyl halide as a radical generator,43–45 led us to consider the use of Cu metal as a reagent in the preparation of alkoxyamines. In this study, we report the synthesis of a small library of alkoxyamines from various alkyl halides and nitroxides in a very simple and efficient way using Cu metal. The reaction is summarized in Scheme 2.
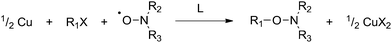 | ||
| Scheme 2 Copper metal-mediated reaction of alkyl halides with nitroxides (L = bipy or PMDETA). | ||
Results and discussion
In an initial investigation, the reaction was performed in a number of different solvents of varying polarity at 30 °C, using ethyl 2-bromoisobutyrate (1 eq.) as the substrate, SG1 (1 eq.) as the nitroxide, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 0.6 eq.) as the ligand and 0.2 mm diameter copper wire (0.5 eq.) as the reducing agent. The formation of alkoxyamine A1 (Table 1) was tracked using 1H NMR spectroscopy (Fig. 1) by comparing the area of the peak corresponding to the methyl groups of ethyl 2-bromoisobutyrate (δ 1.91 ppm, 6H) and the areas of the peaks corresponding to the methyl groups of the SG1 adduct (δ 1.57 and 1.67 ppm, 2 × 3H). The formation of the desired alkoxyamine was observed, which confirms the efficiency of the synthetic pathway employed here.| Expt. | R1–X | R2R3NO˙ | R1–ONR2R3 | Liganda | Solvent | Cu (eq. to R1–X) | Time /h | Isolated yield |
|---|---|---|---|---|---|---|---|---|
| a PMDETA: N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (1.2 eq. relative to Cu); bipy: 2,2′-bipyridine (2 eq. relative to Cu). b See ESI† for the synthetic procedure. c Cu powder was used in place of copper wire. d When PMDETA was used as ligand, no alkoxyamine was formed. | ||||||||
| A1 |

|

|

|
PMDETA | DMSO b | 1c | 1 | 94% |
| PMDETA | DMSO b | 0.5 | 2 | 91% | ||||
| PMDETA | MeCN | 0.5 | 1 | 91% | ||||
| PMDETA | EtOH b | 0.5 | 2 | 85% | ||||
| A2 |

|

|

|
bipy | MeCN | 0.5 | 16 | 89% |
| PMDETA | MeCN b | 0.5 | 6 | 55% | ||||
| A3 |

|

|

|
bipy d | MeCN | 0.5 | 16 | 89% |
| A4 |

|

|

|
bipy | MeCN | 1c | 16 | 87% |
| PMDETA | MeCN b | 1c | 16 | 55% | ||||
| PMDETA | MeCN | 0.5c | 16 | 32% | ||||
| A5 |

|

|

|
PMDETA | MeCN | 1 | 16 | 78%c |
| A6 |

|

|

|
bipy d | MeCN | 0.5 | 16 | 71% |
| A7 |

|

|

|
PMDETA | MeCN | 0.5 | 3 | 87% |
| A8 |

|

|

|
PMDETA | MeCN | 0.5 | 3 | 72% |
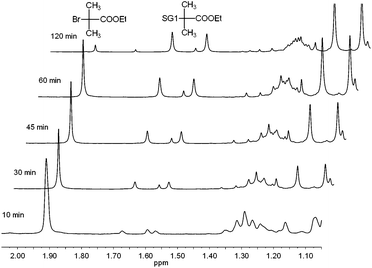 | ||
| Fig. 1 Extracts from 1H NMR spectra (CDCl3, 300 MHz) of the copper-mediated reaction between ethyl 2-bromo-2-methylpropionate and SG1 in DMSO, showing changing intensities of the peaks corresponding to methyl protons of ethyl 2-bromo-2-methylpropionate at 1.91 ppm and the SG1 adduct at 1.57 and 1.67 ppm. | ||
In addition, the reaction was much more rapid in acetonitrile (MeCN), DMSO and ethanol (EtOH) than in ethyl acetate (EtOAc) and toluene, in accordance with previous polymerization results.38–40,46–48 The fastest reaction was observed in MeCN (Fig. 2). This is consistent with polymerization results which show that, at least in the early stages of polymerization, rates of copper-mediated polymerization in MeCN are similar to those in DMSO.47,48MeCN is more volatile than DMSO, and hence easier to remove during the purification step. Therefore it was used for subsequent copper metal-mediated reactions.
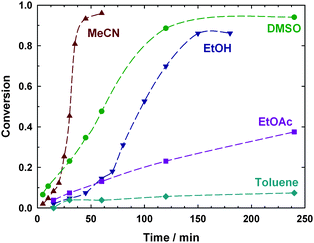 | ||
| Fig. 2 Effect of solvent on the rate of formation of alkoxyamine A1 in the copper-mediated reaction between 1 eq. ethyl 2-bromoisobutyrate and 1 eq. SG1 in the presence of 0.5 eq. Cu wire and 0.6 eq. PMDETA (lines connecting data points are guides for the eye only). | ||
A range of different alkyl halides and nitroxides were employed to determine the versatility of the method and to prepare a small library of alkoxyamines (Table 1). In a typical procedure, activated alkyl bromide (1 eq.) and nitroxide (1.08 eq.) were dissolved in 5 mL acetonitrile. Copper wire (0.2 mm diameter, 0.5 eq.) was added and the solution was sealed and degassed by bubbling with argon. PMDETA (0.6 eq.) was added by syringe. When 2,2′-bipyridine (bipy) was used as a ligand, 1 eq. of bipy was added prior to degassing. The different ratios of bipy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu and PMDETA:Cu reflect the different stoichiometries of the Cu(PMDETA)Br and Cu(bipy)2Br complexes.49 The reaction was stirred at room temperature for 1–16 h, depending on the substrate. While reactions involving the active tertiary substrate, ethyl 2-bromoisobutyrate (see A1 and A7–A8), were complete in 1–3 h, less active primary and secondary substrates were allowed to react for 16 h to ensure full conversion. The reaction mixture was diluted with EtOAc and copper(II) byproducts were removed by washing with dilute HCl, NaHCO3 and brine. The product was separated from any remaining copper and unreacted nitroxide by column chromatography on silica.
:Cu and PMDETA:Cu reflect the different stoichiometries of the Cu(PMDETA)Br and Cu(bipy)2Br complexes.49 The reaction was stirred at room temperature for 1–16 h, depending on the substrate. While reactions involving the active tertiary substrate, ethyl 2-bromoisobutyrate (see A1 and A7–A8), were complete in 1–3 h, less active primary and secondary substrates were allowed to react for 16 h to ensure full conversion. The reaction mixture was diluted with EtOAc and copper(II) byproducts were removed by washing with dilute HCl, NaHCO3 and brine. The product was separated from any remaining copper and unreacted nitroxide by column chromatography on silica.
In some cases, low yields were obtained when PMDETA was used as a ligand (see A2–A4 and A6), possibly due to the formation of quaternary ammonium salts (1° and 2° halides) or base-catalyzed dehydrobromination (2° and 3° halides).50 In these cases, yields were significantly improved by substituting bipy (2 eq. relative to Cu) for PMDETA. With this modification, good isolated yields were obtained across a range of activated primary, secondary and tertiary bromide substrates.
Cu/PMDETA complexes are more active than Cu/bipy complexes in ATRP (KATRP = 7.5 × 10−8 and 3.9 × 10−9 respectively with ethyl 2-bromoisobutyrate in MeCN at 22 °C).51 This difference implies that Cu/PMDETA complexes will convert alkyl halides to radicals more rapidly than Cu/bipy complexes. This in turn should lead to a higher yield of alkoxyamines, the opposite of what was observed. Yields with PMDETA were generally observed to decrease as the substrate became less hindered (3° > 2° > 1° substrates), which is in line with expectations if the chief side reaction is formation of a quaternary ammonium salt with PMDETA, but inconsistent with side reactions such as excessive bimolecular termination caused by too-rapid formation of radicals.
Similar yields of alkoxyamine were obtained when copper powder was used in place of copper wire (see A1). From alkyl chloride substrates such as benzyl chloride and 2-chloropropionic acid, however, very low (≤5%) yields of alkoxyamine were obtained, perhaps due to the higher strength of the C–Cl bond compared to C–Br (data not shown). While increasing the temperature may have improved these yields, the appealing feature of a room temperature reaction would have been lost.
The acid-functionalized substrates, 2-methyl-2-bromopropionic acid (A4, the precursor to BlocBuilder MA) and 2-bromopropionic acid (A5), required the addition of an equimolar amount of copper powder in order to obtain the desired alkoxyamine in 70–90% yield after 16 h. When a stoichiometric amount (0.5 eq.) of copper powder was used in the synthesis of A4, a yield of only 32% was obtained after 16 h. The resulting acid-functionalized alkoxyamines were readily separated by dissolving the reaction mixture in a 1 M solution of NaOH, washing with diethyl ether, then acidifying the aqueous phase and extracting with dichloromethane. In contrast, the amine-functionalized alkoxyamine (A8) was prepared from TEMPO–NH2 in 72% isolated yield after 3 h reaction using a stoichiometric quantity (0.5 eq.) of copper wire. Another TEMPO derivative, 4-oxo-TEMPO, was also used with ethyl 2-bromo-2-methylpropionate to prepare the corresponding alkoxyamine (A7) in 87% yield.
The difficulty encountered in preparing acid-functionalized alkoxyamines may be due to protonation of the ligand (bipy or PMDETA) and its displacement from the copper(I) complex by the carboxylic acids of the substrate. Reaction mixtures containing acid substrates were blue, compared to the green solutions obtained from non-acid substrates, suggesting a change in the structure of the copper complex. While copper(I) acetate bis(4,4′-dinonyl-2,2′-bipyridine) has been shown to be an effective initiator of polymerization,52 acidic monomers such as methacrylic acid cannot be polymerized by ATRP except under basic conditions such that the acid is deprotonated.53 Even at high pH, Ashford et al. observed very low rates of polymerization of sodium methacrylate when using sodium 2-bromo-2-methylpropionate as an initiator.53 In our experiments with acid-functionalized substrates, twice the usual amount of copper metal was required to prepare the corresponding alkoxyamines in good yield, and nearly all of this copper was converted to copper salts (presumably copper(I)). This indicates that the presence of carboxylic acids inhibits the further oxidation of Cu(I) to Cu(II) by activated alkyl halides, probably by displacing the PMDETA or bipy ligands from the copper complex.
Experimental
Materials
All materials were used without further purification. N-tert-Butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide SG1 (85%) was kindly supplied by Arkema. All other reagents were obtained from Sigma-Aldrich at the highest available purity.Instrumentation
1H and 13C NMR spectroscopy were performed in 5 mm diameter tubes in CDCl3 on a Bruker Avance-300 (300 MHz) spectrometer. The chemical shift scale was calibrated on the basis of the solvent peak (δ = 7.26 ppm). Mass spectrometry was performed on a Bruker Esquire-LC instrument using electrospray ionization with MeOH as solvent.Kinetic experiments to determine the effect of solvent on the rate of formation of ethyl 2-methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-di-methylpropyl)-aminoxy] propionate (A1)
SG1 (0.94 g, 2.7 mmol), copper wire (0.2 mm diameter, 80 mg, 1.25 mmol), ethyl 2-bromo-2-methylpropionate (0.49 g, 2.5 mmol) and solvent (DMSO, EtOH, EtOAc, MeCN, or toluene, 5 mL) were added to a 25 mL round bottomed flask with magnetic stirrer bar. The flask was sealed with a rubber septum, placed in an oil bath at 30 °C, and degassed by bubbling with Ar for 10 min while stirring. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA, 0.3 mL, 0.25 g, 1.5 mmol) was added by syringe. The flask was stirred at 30 °C, and samples were removed by syringe at regular intervals. The samples were immediately diluted with CDCl3 and 1H NMR spectra were obtained. Conversion was calculated by comparing the area under the peak at 1.91 ppm (ethyl 2-bromo-2-methylpropionate, (CH3)2CBr, 6H) with the sum of the areas under the peaks at 1.57 and 1.67 ppm (SG1 adduct, (CH3)2CSG1, 2 × 3H) (Fig. 1).Synthesis of alkoxyamines
:1 EtOAc:hexane), total yield 0.936 g (91%). 1H NMR (CDCl3, 300 MHz, 25 °C): δ 1.08 ((CH3)3C, s, 9H), 1.17 ((CH3)3C, s, 9H), 1.21–1.33 ((CH3CH2O)2P + CH3CH2OCO, m, 9H), 1.58 ((CH3)2C, s, 3H), 1.63 ((CH3)2C, s, 3H), 3.26 (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CHPO3Et2, d, 1H, 2JH-P = 25.8 Hz), 3.85–4.45 ((CH3CH2O)2P + CH3CH2OCO, m, 6H). 13C NMR (CDCl3, 75 MHz, 25 °C): δ 13.9 (CH3CH2OCO), 16.0 (CH3CH2OP, d, 3JC-P = 6.4 Hz), 16.4 (CH3CH2OP, d, 3JC-P = 5.8 Hz), 22.3 ((CH3)2C), 27.8 ((CH3)2C), 28.0 ((CH3)3CN), 29.8 ((CH3)3CCHP, d, 3JC-P = 5.3 Hz), 35.7 ((CH3)3CCHP, d, 2JC-P = 6.3 Hz), 58.3 (CH3CH2OP, d, 2JC-P = 7.3 Hz), 60.5 (CH3CH2OCO), 61.5 (CH3CH2OP, d, 2JC-P = 5.9 Hz), 61.9 ((CH3)3CN), 69.9 ((CH3)3CCHP, d, 1JC-P = 137 Hz), 83.4 ((CH3)2CCO2Et), 174.7 ((CH3)2CCO2Et) ppm. MS (ESI): m/z = 432 (M + Na+). Calc. for C19H40NNaO6P: 432.3.
CHPO3Et2, d, 1H, 2JH-P = 26.1 Hz), 3.15–4.05 ((CH3CH2O)2P, m, 4H), 5.24 (CH3CH(Ph)O, q, 1H, J = 6.6 Hz), 7.15–7.50 (C6H5, m, 5H) ppm. Minor diastereomer: δ 0.83 ((CH3)3C, s, 9H), 1.09–1.37 ((CH3CH2O)2P, m, 6H), 1.23 ((CH3)3C, s, 9H), 1.59 (CH3CH(Ph)O, d, 3H, J = 6.8 Hz), 3.34 (CHPO3Et2, d, 1H, 2JH-P = 26.1 Hz), 3.75–4.45 ((CH3CH2O)2P, m, 4H), 4.98 (CH3CH(Ph)O, q, 1H, J = 6.7 Hz), 7.25–7.35 (C6H5, m, 5H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): major diastereomer: 16.3 (CH3CH2OP, d, 3JC-P = 6.3 Hz), 16.7 (CH3CH2OP, d, 3JC-P = 5.5 Hz), 28.2 ((CH3)3CN), 28.6 (CH3CH(Ph)O), 30.6 ((CH3)3CCHP, d, 3JC-P = 5.8 Hz), 35.3 (Me3CCHP, d, 2JC-P = 5.0 Hz), 58.6 (CH3CH2OP, d, 2JC-P = 7.5 Hz), 61.2 (CH3CH2OP), 61.6 (Me3CN), 70.1 (tBuCHP, d, 1JC-P = 139 Hz), 78.3 (MeCH(Ph)O), 127.1 (C6H5, para), 127.9 (C6H5, meta), 126.9 (C6H5, ortho), 143.3 (C6H5, quat.) ppm. Minor diastereomer: 16.3 (CH3CH2OP, d, 3JC-P = 6.3 Hz), 16.7 (CH3CH2OP, d, 3JC-P = 5.5 Hz), 24.1 (CH3CH(Ph)O), 28.5 ((CH3)3CN), 30.1 ((CH3)3CCHP, d, 3JC-P = 5.5 Hz), 35.6 (Me3CCHP, d, 2JC-P = 5.7 Hz), 58.8 (CH3CH2OP, d, 2JC-P = 7.2 Hz), 61.1 (CH3CH2OP), 61.6 (Me3CN), 69.8 (tBuCHP, d, 1JC-P = 139 Hz), 85.3 (MeCH(Ph)O), 127.3 (C6H5, ortho), 127.8 (C6H5, para), 128.0 (C6H5, meta), 145.4 (C6H5, quat.) ppm. MS (ESI): m/z = 422 (M + Na+). Calc. for C21H38NNaO4P: 422.2.
CHPO3Et2, d, 1H, 2JH-P = 16.0 Hz), 3.85–4.30 ((CH3CH2O)2P, m, 4H), 4.48 (OCH2CN, d, 1H, 2JH-H = 15.7 Hz), 4.80 (OCH2CN, d, 1H, 2JH-H = 15.5 Hz). 13C NMR (CDCl3, 75 MHz, 25 °C): 16.2 (CH3CH2OP, d, 3JC-P = 6.7 Hz), 16.6 (CH3CH2OP, d, 3JC-P = 5.3 Hz), 27.5 ((CH3)3CN), 29.7 ((CH3)3CCHP, d, 3JC-P = 5.2 Hz), 35.3 (Me3CCHP, d, 2JC-P = 4.1 Hz), 59.5 (CH3CH2OP, d, 2JC-P = 7.0 Hz), 61.5 (Me3CN), 61.7 (CH3CH2OP, d, 2JC-P = 6.4 Hz), 62.5 (OCH2CN), 69.5 (tBuCHP, d, 1JC-P = 140 Hz), 115.6 (OCH2CN) ppm. MS (ESI): m/z = 357 (M + Na+). Calc. for C15H31N2NaO4P: 357.2.
CHPO3Et2, d, 1H, 2JH-P = 26.4 Hz), 3.95–4.40 ((CH3CH2O)2P, m, 4H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): δ 16.1 (CH3CH2OP, d, 3JC-P = 6.6 Hz), 16.5 (CH3CH2OP, d, 3JC-P = 5.4 Hz), 24.3 ((CH3)2C), 28.3 ((CH3)2C), 28.7 ((CH3)3CN), 29.8 ((CH3)3CCHP, d, 3JC-P = 5.4 Hz), 36.1 (Me3CCHP, d, 2JC-P = 5.9 Hz), 60.3 (CH3CH2OP, d, 2JC-P = 7.7 Hz), 62.4 (CH3CH2OP, d, 2JC-P = 6.6 Hz), 63.3 (Me3CN), 69.9 (tBuCHP, d, 1JC-P = 137 Hz), 84.8 (Me2CCOOH), 176.8 (Me2CCOOH) ppm. MS (ESI): m/z = 382 (M + H+). Calc. for C17H37NO6P: 382.2.
CHPO3Et2, d, 1H, 2JH-P = 25.7 Hz), 3.90–4.28 ((CH3CH2O)2P, m, 4H), 4.60 (CH3CHCOOH, q, 1H, J = 6.9 Hz) ppm. Minor diastereomer: δ 1.12 ((CH3)3C, s, 9H), 1.17 ((CH3)3C, s, 9H), 1.21–1.38 ((CH3CH2O)2P, m, 6H), 1.49 (CH3CHCOOH, d, 3H, 3JH-H = 7.0 Hz), 3.37 (CHPO3Et2, d, 1H, 2JH-P = 27.5 Hz), 3.96–4.36 ((CH3CH2O)2P, m, 4H), 4.54 (CH3CHCOOH, q, 1H, J = 7.0 Hz) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): major diastereomer: 16.1 (CH3CH2OP, d, 3JC-P = 5.7 Hz), 16.3 (CH3CH2OP, d, 3JC-P = 4.5 Hz), 18.5 (CH3CHCOOH), 27.7 ((CH3)3CN), 30.2 ((CH3)3CCHP, d, 3JC-P = 5.7 Hz), 35.5 (Me3CCHP, d, 2JC-P = 4.5 Hz), 60.6 (CH3CH2OP, d, 2JC-P = 7.8 Hz), 62.5 (CH3CH2OP, d, 2JC-P = 7.4 Hz), 62.5 (Me3CN), 68.1 (tBuCHP, d, 1JC-P = 139 Hz), 81.4 (MeCHCOOH), 174.3 (MeCHCOOH) ppm. Minor diastereomer: 16.0 (CH3CH2OP, d, 3JC-P = 6.0 Hz), 16.3 (CH3CH2OP, d, 3JC-P = 4.8 Hz), 18.9 (CH3CHCOOH), 27.8 ((CH3)3CN), 29.6 ((CH3)3CCHP, d, 3JC-P = 5.6 Hz), 35.5 (Me3CCHP, d, 2JC-P = 4.5 Hz), 59.7 (CH3CH2OP, d, 2JC-P = 7.4 Hz), 62.2 (CH3CH2OP, d, 2JC-P = 7.8 Hz), 62.2 (Me3CN), 69.5 (CH3CH2OP, d, 1JC-P = 141 Hz), 82.3 (MeCHCOOH), 175.4 (MeCHCOOH) ppm. MS (ESI): m/z = 368 (M + H+). Calc. for C16H35NO6P: 368.2.
CHPO3Et2, d, 1H, 2JH-P = 24.4 Hz), 3.4–4.1 ((CH3CH2O)2P, m, 4H), 4.58 (PhCH2O, d, 1H, J = 9.4 Hz), 5.23 (PhCH2O, d, 1H, J = 9.5 Hz), 7.2–7.5 (C6H5, m, 5H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): 16.2 (CH3CH2OP, d, 3JC-P = 6.8 Hz), 16.4 (CH3CH2OP, d, 3JC-P = 5.8 Hz), 27.8 ((CH3)3CN), 30.1 ((CH3)3CCHP, d, 3JC-P = 5.9 Hz), 35.3 (Me3CCHP, d, 2JC-P = 5.3 Hz), 58.8 (CH3CH2OP, d, 2JC-P = 7.2 Hz), 61.6 (CH3CH2OP, d, 2JC-P = 6.4 Hz), 62.0 (Me3CN), 69.7 (CH3CH2OP, d, 1JC-P = 140 Hz), 77.8 (PhCH2O), 127.7 (C6H5, para), 128.0 (C6H5, meta), 129.6 (C6H5, ortho), 136.8 (C6H5, quat.) ppm. MS (ESI): m/z = 408 (M + Na+). Calc. for C20H36NNaO4P: 408.2.
CHPO3Et2, d, 1H, 2JH-P = 25.8 Hz), 3.85–4.45 ((CH3CH2O)2P + CH3CH2OCO, m, 6H). 13C NMR (CDCl3, 75 MHz, 25 °C): δ 13.9 (CH3CH2OCO), 16.0 (CH3CH2OP, d, 3JC-P = 6.4 Hz), 16.4 (CH3CH2OP, d, 3JC-P = 5.8 Hz), 22.3 ((CH3)2C), 27.8 ((CH3)2C), 28.0 ((CH3)3CN), 29.8 ((CH3)3CCHP, d, 3JC-P = 5.3 Hz), 35.7 ((CH3)3CCHP, d, 2JC-P = 6.3 Hz), 58.3 (CH3CH2OP, d, 2JC-P = 7.3 Hz), 60.5 (CH3CH2OCO), 61.5 (CH3CH2OP, d, 2JC-P = 5.9 Hz), 61.9 ((CH3)3CN), 69.9 ((CH3)3CCHP, d, 1JC-P = 137 Hz), 83.4 ((CH3)2CCO2Et), 174.7 ((CH3)2CCO2Et) ppm. MS (ESI): m/z = 432 (M + Na+). Calc. for C19H40NNaO6P: 432.3.
CHPO3Et2, d, 1H, 2JH-P = 26.1 Hz), 3.15–4.05 ((CH3CH2O)2P, m, 4H), 5.24 (CH3CH(Ph)O, q, 1H, J = 6.6 Hz), 7.15–7.50 (C6H5, m, 5H) ppm. Minor diastereomer: δ 0.83 ((CH3)3C, s, 9H), 1.09–1.37 ((CH3CH2O)2P, m, 6H), 1.23 ((CH3)3C, s, 9H), 1.59 (CH3CH(Ph)O, d, 3H, J = 6.8 Hz), 3.34 (CHPO3Et2, d, 1H, 2JH-P = 26.1 Hz), 3.75–4.45 ((CH3CH2O)2P, m, 4H), 4.98 (CH3CH(Ph)O, q, 1H, J = 6.7 Hz), 7.25–7.35 (C6H5, m, 5H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): major diastereomer: 16.3 (CH3CH2OP, d, 3JC-P = 6.3 Hz), 16.7 (CH3CH2OP, d, 3JC-P = 5.5 Hz), 28.2 ((CH3)3CN), 28.6 (CH3CH(Ph)O), 30.6 ((CH3)3CCHP, d, 3JC-P = 5.8 Hz), 35.3 (Me3CCHP, d, 2JC-P = 5.0 Hz), 58.6 (CH3CH2OP, d, 2JC-P = 7.5 Hz), 61.2 (CH3CH2OP), 61.6 (Me3CN), 70.1 (tBuCHP, d, 1JC-P = 139 Hz), 78.3 (MeCH(Ph)O), 127.1 (C6H5, para), 127.9 (C6H5, meta), 126.9 (C6H5, ortho), 143.3 (C6H5, quat.) ppm. Minor diastereomer: 16.3 (CH3CH2OP, d, 3JC-P = 6.3 Hz), 16.7 (CH3CH2OP, d, 3JC-P = 5.5 Hz), 24.1 (CH3CH(Ph)O), 28.5 ((CH3)3CN), 30.1 ((CH3)3CCHP, d, 3JC-P = 5.5 Hz), 35.6 (Me3CCHP, d, 2JC-P = 5.7 Hz), 58.8 (CH3CH2OP, d, 2JC-P = 7.2 Hz), 61.1 (CH3CH2OP), 61.6 (Me3CN), 69.8 (tBuCHP, d, 1JC-P = 139 Hz), 85.3 (MeCH(Ph)O), 127.3 (C6H5, ortho), 127.8 (C6H5, para), 128.0 (C6H5, meta), 145.4 (C6H5, quat.) ppm. MS (ESI): m/z = 422 (M + Na+). Calc. for C21H38NNaO4P: 422.2.
CHPO3Et2, d, 1H, 2JH-P = 16.0 Hz), 3.85–4.30 ((CH3CH2O)2P, m, 4H), 4.48 (OCH2CN, d, 1H, 2JH-H = 15.7 Hz), 4.80 (OCH2CN, d, 1H, 2JH-H = 15.5 Hz). 13C NMR (CDCl3, 75 MHz, 25 °C): 16.2 (CH3CH2OP, d, 3JC-P = 6.7 Hz), 16.6 (CH3CH2OP, d, 3JC-P = 5.3 Hz), 27.5 ((CH3)3CN), 29.7 ((CH3)3CCHP, d, 3JC-P = 5.2 Hz), 35.3 (Me3CCHP, d, 2JC-P = 4.1 Hz), 59.5 (CH3CH2OP, d, 2JC-P = 7.0 Hz), 61.5 (Me3CN), 61.7 (CH3CH2OP, d, 2JC-P = 6.4 Hz), 62.5 (OCH2CN), 69.5 (tBuCHP, d, 1JC-P = 140 Hz), 115.6 (OCH2CN) ppm. MS (ESI): m/z = 357 (M + Na+). Calc. for C15H31N2NaO4P: 357.2.
CHPO3Et2, d, 1H, 2JH-P = 26.4 Hz), 3.95–4.40 ((CH3CH2O)2P, m, 4H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): δ 16.1 (CH3CH2OP, d, 3JC-P = 6.6 Hz), 16.5 (CH3CH2OP, d, 3JC-P = 5.4 Hz), 24.3 ((CH3)2C), 28.3 ((CH3)2C), 28.7 ((CH3)3CN), 29.8 ((CH3)3CCHP, d, 3JC-P = 5.4 Hz), 36.1 (Me3CCHP, d, 2JC-P = 5.9 Hz), 60.3 (CH3CH2OP, d, 2JC-P = 7.7 Hz), 62.4 (CH3CH2OP, d, 2JC-P = 6.6 Hz), 63.3 (Me3CN), 69.9 (tBuCHP, d, 1JC-P = 137 Hz), 84.8 (Me2CCOOH), 176.8 (Me2CCOOH) ppm. MS (ESI): m/z = 382 (M + H+). Calc. for C17H37NO6P: 382.2.
CHPO3Et2, d, 1H, 2JH-P = 25.7 Hz), 3.90–4.28 ((CH3CH2O)2P, m, 4H), 4.60 (CH3CHCOOH, q, 1H, J = 6.9 Hz) ppm. Minor diastereomer: δ 1.12 ((CH3)3C, s, 9H), 1.17 ((CH3)3C, s, 9H), 1.21–1.38 ((CH3CH2O)2P, m, 6H), 1.49 (CH3CHCOOH, d, 3H, 3JH-H = 7.0 Hz), 3.37 (CHPO3Et2, d, 1H, 2JH-P = 27.5 Hz), 3.96–4.36 ((CH3CH2O)2P, m, 4H), 4.54 (CH3CHCOOH, q, 1H, J = 7.0 Hz) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): major diastereomer: 16.1 (CH3CH2OP, d, 3JC-P = 5.7 Hz), 16.3 (CH3CH2OP, d, 3JC-P = 4.5 Hz), 18.5 (CH3CHCOOH), 27.7 ((CH3)3CN), 30.2 ((CH3)3CCHP, d, 3JC-P = 5.7 Hz), 35.5 (Me3CCHP, d, 2JC-P = 4.5 Hz), 60.6 (CH3CH2OP, d, 2JC-P = 7.8 Hz), 62.5 (CH3CH2OP, d, 2JC-P = 7.4 Hz), 62.5 (Me3CN), 68.1 (tBuCHP, d, 1JC-P = 139 Hz), 81.4 (MeCHCOOH), 174.3 (MeCHCOOH) ppm. Minor diastereomer: 16.0 (CH3CH2OP, d, 3JC-P = 6.0 Hz), 16.3 (CH3CH2OP, d, 3JC-P = 4.8 Hz), 18.9 (CH3CHCOOH), 27.8 ((CH3)3CN), 29.6 ((CH3)3CCHP, d, 3JC-P = 5.6 Hz), 35.5 (Me3CCHP, d, 2JC-P = 4.5 Hz), 59.7 (CH3CH2OP, d, 2JC-P = 7.4 Hz), 62.2 (CH3CH2OP, d, 2JC-P = 7.8 Hz), 62.2 (Me3CN), 69.5 (CH3CH2OP, d, 1JC-P = 141 Hz), 82.3 (MeCHCOOH), 175.4 (MeCHCOOH) ppm. MS (ESI): m/z = 368 (M + H+). Calc. for C16H35NO6P: 368.2.
CHPO3Et2, d, 1H, 2JH-P = 24.4 Hz), 3.4–4.1 ((CH3CH2O)2P, m, 4H), 4.58 (PhCH2O, d, 1H, J = 9.4 Hz), 5.23 (PhCH2O, d, 1H, J = 9.5 Hz), 7.2–7.5 (C6H5, m, 5H) ppm. 13C NMR (CDCl3, 75 MHz, 25 °C): 16.2 (CH3CH2OP, d, 3JC-P = 6.8 Hz), 16.4 (CH3CH2OP, d, 3JC-P = 5.8 Hz), 27.8 ((CH3)3CN), 30.1 ((CH3)3CCHP, d, 3JC-P = 5.9 Hz), 35.3 (Me3CCHP, d, 2JC-P = 5.3 Hz), 58.8 (CH3CH2OP, d, 2JC-P = 7.2 Hz), 61.6 (CH3CH2OP, d, 2JC-P = 6.4 Hz), 62.0 (Me3CN), 69.7 (CH3CH2OP, d, 1JC-P = 140 Hz), 77.8 (PhCH2O), 127.7 (C6H5, para), 128.0 (C6H5, meta), 129.6 (C6H5, ortho), 136.8 (C6H5, quat.) ppm. MS (ESI): m/z = 408 (M + Na+). Calc. for C20H36NNaO4P: 408.2.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ring)) ppm. MS (ESI): m/z = 308 (M + Na+). Calc. for C15H27NNaO4: 308.2.
O (ring)) ppm. MS (ESI): m/z = 308 (M + Na+). Calc. for C15H27NNaO4: 308.2.
Conclusions
A simple and efficient method has been developed for the preparation of alkoxyamines, including thermally labile SG1-based alkoxyamines such as BlocBuilder MA, which has been demonstrated to be particularly efficient for the control of radical polymerizations. This method is compatible with a wide range of functionalities, including acid and amine groups. A stoichiometric amount of copper metal is used as a reducing agent, with no requirement for addition of copper(I) or copper(II) salts at the start of the reaction. The reaction proceeds rapidly at room temperature in polar solvents such as MeCN, EtOH or DMSO. Copper wire is a cheap and convenient reducing agent, whose dissolution provides a useful visual indication of the progress of the reaction. Purification is straightforward, and isolated yields of 71–94% are obtained across a wide range of activated alkyl bromide and nitroxide substrates.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Community's Seventh Framework Programme FP7/2007-2013 (Grant Agreement No. 249835). The authors are grateful to Arkema for kindly providing the SG1 nitroxide. CNRS and French ministry of research are warmly acknowledged for funding.Notes and references
- D. Crich, D. Grant and A. A. Bowers, J. Am. Chem. Soc., 2007, 129, 12106–12107 Search PubMed.
- A. Studer and T. Schulte, Chem. Rec., 2005, 5, 27–35 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J. P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- F. Chauvin, P. E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34–47 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. P. Magnet, Angew. Chem., Int. Ed., 2004, 43, 6186–6189 CrossRef.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639–3647 CrossRef CAS.
- T. Trimaille, K. Mabrouk, V. Monnier, L. Charles, D. Bertin and D. Gigmes, Macromolecules, 2010, 43, 4864–4870 CrossRef CAS.
- M. Chenal, C. Boursier, Y. Guillaneuf, M. Taverna, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2 10.1039/c1py00028d.
- M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291–9303 CrossRef CAS.
- P. Nesvadba, Chimia, 2006, 60, 832–840 CrossRef CAS.
- A. C. Greene and R. B. Grubbs, in ACS Symp. Ser., ed. K. Matyjaszewski, American Chemical Society, Washington DC, 2009, vol. 1024 Search PubMed.
- D. Wang and Z. Wu, Macromolecules, 1998, 31, 6727–6729 Search PubMed.
- Y. Guillaneuf, J. L. Couturier, D. Gigmes, S. R. A. Marque, P. Tordo and D. Bertin, J. Org. Chem., 2008, 73, 4728–4731 CrossRef CAS.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955–5957 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. Marchand, L. Autissier, Y. Guillaneuf, J. L. Couturier, D. Gigmes and D. Bertin, Aust. J. Chem., 2010, 63, 1237–1244 Search PubMed.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Macromolecules, 2005, 38, 2638–2650 CrossRef CAS.
- D. Bertin, D. Gigmes, C. Le Mercier, S. R. A. Marque and P. Tordo, J. Org. Chem., 2004, 69, 4925–4930 CrossRef CAS.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D. L. Versace, J. Lalevee and J. P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- S. Miele, P. Nesvadba and A. Studer, Macromolecules, 2009, 42, 2419–2427 CrossRef CAS.
- J. Ruehl, N. Ningnuek, T. Thongpaisanwong and R. Braslau, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8049–8069 Search PubMed.
- N. D. Contrella and E. S. Tillman, Polymer, 2008, 49, 4076–4079 Search PubMed.
- R. B. Grubbs, J. K. Wegrzyn and Q. Xia, Chem. Commun., 2005, 80–82 RSC.
- A. C. Greene and R. B. Grubbs, Macromolecules, 2009, 42, 4388–4390 CrossRef CAS.
- A. C. Greene and R. B. Grubbs, Macromolecules, 2010, 43, 10320–10325 CrossRef CAS.
- J. Kulis, C. A. Bell, A. S. Micallef, Z. F. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218–8227 CrossRef CAS.
- J. Kulis, C. A. Bell, A. S. Micallef and M. J. Monteiro, Aust. J. Chem., 2010, 63, 1227–1236 Search PubMed.
- J. Kulis, C. A. Bell, A. S. Micallef and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2214–2223 CrossRef CAS.
- Y. G. Li, Y. Q. Zhang, D. Yang, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2084–2097 Search PubMed.
- W. C. Lin, B. Huang, Q. A. Fu, G. W. Wang and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2991–2999 CrossRef CAS.
- G. W. Wang, Y. N. Zhang and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1633–1640 Search PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- B. D. Hornby, A. G. West, J. C. Tom, C. Waterson, S. Harrisson and S. Perrier, Macromol. Rapid Commun., 2010, 31, 1276–1280 Search PubMed.
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS.
- J. Tom, B. Hornby, A. West, S. Harrisson and S. Perrier, Polym. Chem., 2010, 1, 420–422 RSC.
- X. H. Liu, G. B. Zhang, B. X. Li, Y. G. Bai and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5439–5445 Search PubMed.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4889–4893 Search PubMed.
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. L. Wei and B. E. Woodworth, Macromolecules, 1997, 30, 7348–7350 CrossRef CAS.
- V. Percec, B. Barboiu and M. van der Sluis, Macromolecules, 1998, 31, 4053–4056 CrossRef CAS.
- M. van der Sluis, B. Barboiu, N. Pesa and V. Percec, Macromolecules, 1998, 31, 9409–9412 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 Search PubMed.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Coord. Chem. Rev., 2005, 249, 1155–1184 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edn, John Wiley and Sons, Inc., New York, 2001 Search PubMed.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS.
- K. Matyjaszewski, M. L. Wei, J. H. Xia and S. G. Gaynor, Macromol. Chem. Phys., 1998, 199, 2289–2292 Search PubMed.
- E. J. Ashford, V. Naldi, R. O'Dell, N. C. Billingham and S. P. Armes, Chem. Commun., 1999, 1285–1286 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for remaining experiments referenced in Table 1. See DOI: 10.1039/c1py00131k |
| This journal is © The Royal Society of Chemistry 2011 |