Inverse thermally reversible gelation-based hydrogels: synthesis and characterization of N-isopropylacrylamide copolymers containing deoxycholic acid in the side chain
Yan
Lu
*a,
Yuqiao
Han
b,
Jinhuan
Liang
b,
Hongxia
Meng
b,
Fengli
Han
a,
Xudong
Wang
b and
Chenxi
Li
*b
aSchool of Materials Science & Engineering, Tianjin University of Technology, Tianjin, 300384, China. E-mail: luyan.tjut@yahoo.com.cn; Fax: +86-22-60215553; Tel: +86-22-60215746
bKey Laboratory of Functional Polymer Materials, Ministry of Education, Institute of Polymer Chemistry, Nankai University, Tianjin, 300071, China. E-mail: nkulicx@yahoo.com.cn; Fax: +86-22-23502749; Tel: +86-22-23501921
First published on 31st May 2011
Abstract
A series of water-soluble, thermosensitive copolymers based on N-isopropylacrylamide (NIPAM) and methacrylate derivatives of deoxycholic acid (MEDCA) were synthesized using free radical copolymerization method by varying feed ratios of monomers. The composition ratios and structure of copolymers were determined by 1H NMR spectroscopy. The glass transition temperature was examined by DSC. The thermoresponsive behaviors of polymeric solutions were investigated by turbidity measurement using UV-Visible spectroscopy. The resultant copolymers exhibit systematic changes in their LCSTs as a function of their chemical composition, as the incorporation of amphiphilic comonomers MEDCA results in a lower and broader LCST of the copolymer solution. An aqueous solution of the copolymer above a critical concentration (2 wt%) experiences four distinct phases such as clear solutions, cloud solutions, gel and shrunken gel upon heating. Finally, the mechanism of the phase transitions was tentatively discussed based on the observations.
Introduction
Aqueous polymer solutions that are transformed into gels by changing the temperature, thus resulting in in situ hydrogel formation, have recently attracted the intensive attention of many investigators for scientific interest and for practical biomedical or pharmaceutical applications.1,2 Among the thermally sensitive polymers, poly(N-isopropylacrylamide), abbreviated as PNIPAM, has stirred up particular interest, chiefly due to its sharp transition behavior and well-defined lower critical solution temperature (LCST) in aqueous medium of around 32 °C which is close to body temperature.3Polymer chains undergo a reversible dehydration and hydration upon heating and cooling, resulting in a coil-to-globule transition.4 The LCST of aqueous PNIPAM solutions can be easily modified by copolymerization with more hydrophilic or more hydrophobic comonomers5 or by addition of salts6 or surfactants.7,8 The related polymers have been widely applied in biomedical and pharmaceutical fields, such as nonviral vector for gene delivery,9 smart culture surface for cell adhesion,10in situ-formable scaffolds,11 and so on.Cholic acid (CA) is a biogenic organic compound synthesized from the cholesterol in the liver of mammals. It is known for its facial amphiphilicity with nonpolar groups on the one side of the steroid skeleton and polar hydroxyl groups and carboxylic group located on the other side.12 The incorporation of CA into polymers may improve the polymer's properties such as thermal properties,13 biodegradability,14 biocompatibility,15,16 and so forth. A large number of reports on the synthesis and characterization of polymeric materials containing cholic acid ester have been reviewed.17
This contribution is a continuation of our work on development new hydrogel systems based on cholic acid-containing polymer.18 In the present work, the deoxycholic acid (DCA), where the 7-position OH group within the cholic acid skeleton was substituted by a H group (Scheme 1), was selected to copolymerize with NIPAM, aiming to develop a novel type of thermoresponsive hydrogel combining the amphiphilic and self-assembling character of DCA with the thermosensitive properties of PNIPAM.
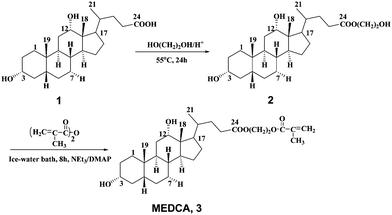 | ||
| Scheme 1 Synthesis of the comonomer derived from deoxycholic acid. | ||
Herein, a series of NIPAM linear random copolymers containing various contents of deoxycholic acid ester derivative (1–5 mol%) were synthesized by conventional free radical polymerization. The chemical composition in the copolymers was determined by NMR spectroscopy. The lower critical solution temperatures (LCSTs) of the polymer solutions were measured by turbidimetry. Very interestingly, different from the NIPAM copolymers with cholic acid in the side chain, aqueous solutions of some of these copolymers above a critical concentration (2 wt%) experience four distinct phases, that is, clear solution, cloudy solution, gel and shrunken gel upon heating, which is relatively rare and has only been reported for poly(NIPAM-acrylic acid)19 and some modified polysaccharides such as an aqueous solution of methyl cellulose.20 The mechanism of the inverse thermally reversible gelation was discussed.
Experimental section
Materials and instruments
N-isopropylacrylamide (NIPAM, Aldrich) was purified by recrystallization in hexane and dried in vacuo at 25 °C. Deoxycholic acid (DCA), N,N-dimethylaminopyridine (DMAP), ethylene glycol and triethylamine were purchased from Acros Organics and were used as received. 2,2′-Azoisobutyronitrile (AIBN, Fluka) was twice recrystallized from methanol and dried in vacuo. Methacrylic anhydride was synthesized according to the literature procedure.21Tetrahydrofuran (THF) dried with sodium wire was used after distillation.The nuclear magnetic resonance (NMR) spectra were recorded on a Varian UNITY Plus-400 NMR spectrometer using CDCl3 as a solvent. All chemical shifts are relative to tetramethylsilane (TMS) set at 0 ppm. The average molecular weights and the distributions of the copolymers were determined by gel permeation chromatography (GPC, Waters-410) using THF as a mobile phase with a flow rate of 1 mL min−1. Monodisperse polystyrene (Polymer Laboratories Inc., MA) was used for calibration. The glass transition temperature (Tg) was recorded on a NETZSCH DSC 204. The LCSTs were determined on a Cary WinUV 100. The phase transition behavior of the polymer solutions was recorded by Nikon E990 digital camera.
Synthesis of (2′-hydroxyethylene)-3α,12α-dihydroxy-5β-deoxycholanoate (2)
Deoxycholic acid (3.0 g, 7.6 mmol) was added into 25 mL of ethylene glycol in a 50 mL round-bottom flask. The mixture was heated to 55 °C with stirring. Concentrated hydrochloride was added dropwise to acidify the solution to pH 3. The reaction mixture was stirred at 55 °C for 24 h, cooled to room temperature, and then poured into water. The white precipitates were filtered and thoroughly washed with water. After drying in a vacuum oven at 60 °C for 24h, 3.1 g of product 2 was obtained with a yield of 93.2%. 1H NMR (400MHz, CDCl3): δ (ppm) = 0.69 (s, 3H, 18-C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 0.91 (s, 3H, 19-C3), 1.00 (d, 3H, 21-C3), 3.63 (m, 1H, 3–O), 3.82 (m, 2H, COOCH2C2OR), 3.97 (s, 1H, 12–O), 4.33 (t, 2H, COOC2CH2OR). Anal. Calcd. for C26H44O5: C, 71.52; H, 10.16. Found: C, 71.37; H, 10.23.
3), 0.91 (s, 3H, 19-C3), 1.00 (d, 3H, 21-C3), 3.63 (m, 1H, 3–O), 3.82 (m, 2H, COOCH2C2OR), 3.97 (s, 1H, 12–O), 4.33 (t, 2H, COOC2CH2OR). Anal. Calcd. for C26H44O5: C, 71.52; H, 10.16. Found: C, 71.37; H, 10.23.
Synthesis of (2′-methacryloyloxyethylene)-3α,12α-dihydroxy-5β-deoxycholanoate (MEDCA, 3)
Compound 2 (3.1 g, 7.1 mmol), triethylamine (3.4 g, 33.8 mmol) and DMAP (50 mg) were dissolved in 50mL of dried THF and cooled in an ice-water bath. Methacrylic anhydride (1.7 g, 10.7 mmol) dissolved in 25 mL of dried THF was added dropwise over a period of 1.5 h. The reaction mixture was stirred for 8 h in ice-water bath, and then the solvent was removed under reduced pressure. The residue was dissolved in a mixture of ethyl acetate and chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and washed with plenty of saturated NaCl solution. The organic layer containing the product was collected, dried with anhydrous MgSO4 and purified by column chromatograph on silica gel (100–200 mesh) with ethyl acetate/petroleum ether (2:1, v/v) as fluent. 1.1g of the product 3 was obtained with a yield of 30.0%. 1H NMR (400MHz, CDCl3): δ (ppm) = 0.67 (s, 3H, 18-C3), 0.91 (s, 3H, 19-C3), 1.01 (d, 3H, 21-C3), 3.64 (m, 1H, 3-O), 3.82 (m, 2H, COOCH2C2OH), 3.98 (s, 1H, 12-O), 4.33 (t, 2H, COOC2CH2OH), 5.60 (s, 1H, C
:1, v/v) and washed with plenty of saturated NaCl solution. The organic layer containing the product was collected, dried with anhydrous MgSO4 and purified by column chromatograph on silica gel (100–200 mesh) with ethyl acetate/petroleum ether (2:1, v/v) as fluent. 1.1g of the product 3 was obtained with a yield of 30.0%. 1H NMR (400MHz, CDCl3): δ (ppm) = 0.67 (s, 3H, 18-C3), 0.91 (s, 3H, 19-C3), 1.01 (d, 3H, 21-C3), 3.64 (m, 1H, 3-O), 3.82 (m, 2H, COOCH2C2OH), 3.98 (s, 1H, 12-O), 4.33 (t, 2H, COOC2CH2OH), 5.60 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2), 6.13 (s, 1H, CC2). Anal. Calcd. for C30H48O6: C, 71.39; H, 9.59. Found: C, 71.35; H, 9.60.
C2), 6.13 (s, 1H, CC2). Anal. Calcd. for C30H48O6: C, 71.39; H, 9.59. Found: C, 71.35; H, 9.60.
Preparation of copolymers
The copolymers of NIPAM and MEDCA, poly(NIPAM-co-MEDCA, were prepared by free radical polymerization in THF with AIBN as the initiator.22 The copolymers containing 1–5 mol% of MEDCA in the feed prior to polymerization were successfully synthesized. The typical polymerization procedure was as follows. The solution of monomers NIPAM and MEDCA in dried THF was purged with nitrogen for about 30 min. Then AIBN (1 mol%) was added into the solution. The temperature of polymerization mixture was raised gradually to 68 °C and maintained for 24 h. The solvents were evaporated and the resulting residue was dissolved in THF, precipitated in diethyl ether and dried in a vacuum oven at 60 °C for 24 h.Glass transition determination
T gs of the copolymers was determined by differential scan calorimetry (DSC) (NETZSCH DSC 204). Each polymer sample (about 12 mg) was heated from ambient temperature to 200 °C at a rate of 10 °C min−1. Tg considered at the mid-point temperature of the exothermic drift in the heating curves was obtained from a second heating after quenching.Thermosensitivity test
LCSTs of all the copolymers were measured by cloud point (turbidity) measurements. The copolymer solutions were prepared in distilled water in an ice-water bath with the concentration of 2.0 wt%. The optical transmittances of these solutions were measured at 500 nm wavelength using UV-vis spectrometer (Cary WinUV 100) equipped with a thermostatic cell with increasing solution temperature (10–40 °C, 0.5 °C min−1). Values for the LCST of copolymer solutions were defined as the temperature at which the transmittance becomes 50% of the initial value at 10 °C.Phase transition temperatures determination
The gelation and the reversibility in the gelation were examined by a vial inversion or tilt method as increasing or decreasing temperature.19 The copolymer solutions (2.0 wt%, 2 mL) in a glass vial (5 mL vol., 1.65 cm dia.) were kept in a thermostated water bath for 5 h prior to inverting the vials. The experiment was carried out at 1 °C intervals. The gelation temperature (Tgel) was visually determined when the polymer solutions did not flow by inverting the vials. In the experiment for observing the shrinkage of gel, the swollen gel formed at Tgel in the glass vial was kept in a thermostated water bath for 10 min prior to tilting the vials. The gel-shrinking temperature (Ts) was also visually determined when the intact gel began to expel water slowly to form a shrunken mass by a vial tilt method.Results and discussion
Synthesis of monomer and copolymers
Methylacrylate monomer was prepared by attaching a polymerizable methacrylic group to the 1,2-ethanediol spacer coupled to the carboxylic acid group at the C-24 position of the deoxycholic acid moiety (Scheme 1). The intermediate (2) was obtained in a high yield (93.2%) by the reaction of deoxycholic acid and ethylene glycol, the later served as both a reactant and a solvent. It seemed that the pH of solution was important in the synthesis. We found the optimal pH value of solution was between 3 and 4. The incorporation of the ethanediol spacer group was expected not only to facilitate the polymerization but also to increase the hydrophilicity of the resultant polymer. The reaction of 2 with methacrylic anhydride in dried THF at lower temperature (ice-water bath) gave the comonomer 3 (MEDCA) in 30% yield.The polymerization of NIPAM with comonomer MEDCA was easily performed in dried THF by free radical polymerization using AIBN as initiator (Copolymers chemical structures see also Scheme 2). A series of copolymers containing deoxycholic acid ester with various chemical compositions were successfully prepared in high yield (80–90%). According to the molar content of MEDCA in feed from 1 to 5 mol%, the prepared copolymers were abbreviated as P1–P5, respectively. It seemed that the extended reaction time was needed, most likely due to the slow kinetics in the polymerization of the derivatives of cholic acid.23
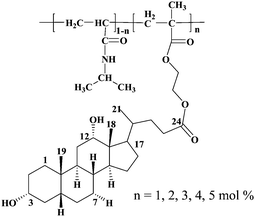 | ||
| Scheme 2 The chemical structure of the deoxycholic acid-containing copolymers. | ||
Characterization of copolymers
The disappearance of the olefinic proton peaks at 5.6 to 6.2 ppm of the monomers in the 1H NMR indicated the successful polymerization of the monomers. The chemical compositions of the copolymers were determined by 1H NMR spectroscopy in CDCl3. The 1H NMR spectra of poly(NIPAM-co-MEDCA) samples were presented in Fig. 1. As shown in Fig. 1, the peaks at 0.68, 0.92 and 1.01 ppm belong to the methyl protons at positions 18, 19 and 21 of the deoxycholic acid skeleton within methylacrylate comonomer, respectively, increasing steadily with the content of the comonomer MEDCA increases in the copolymers. The peaks at 1.15 and 4.02 ppm correspond to the methyl and methane protons of the isopropyl group within NIPAM, respectively. The integration of the characteristic peaks can be used to calculate their chemical compositions.23 Herein, the composition of the copolymers was determined by the integration ratio of peak area of the proton signals from –CH within isopropyl group relative to that of the proton signals from the methyl group at position 18 of the deoxycholic acid skeleton in the copolymer. The calculated results presented in Table 1 show that the chemical compositions of the copolymers are very close to the original monomer compositions in the feed prior to polymerization.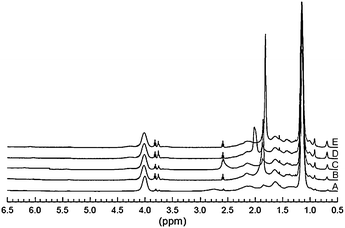 | ||
| Fig. 1
1H NMR spectra of the poly(NIPAM-co-MEDCA) in CDCl3 at room temperature. Molar ratios of NIPAM to MEDCA in feed: (A) 99:1; (B) 98:2; (C) 97:3; (D) 96:4; (E) 95:5. | ||
| Sample code | Molar ratio in the feed | Molar ratio in the polymera | Yield (%) | M w b | PDIb | T g c°C |
|---|---|---|---|---|---|---|
| a Calculated by 1H NMR. b Determined by GPC using THF as a mobile phase. c Determined by DSC. | ||||||
| P1 | 99.0: 1.0 | 99.0: 1.0 | 90 | 15500 | 2.45 | 131.3 |
| P2 | 98.0: 2.0 | 97.8: 2.2 | 86 | 7200 | 2.50 | 120.5 |
| P3 | 97.0: 3.0 | 96.7: 3.3 | 84 | 9100 | 2.58 | 119.7 |
| P4 | 96.0: 4.0 | 95.8: 4.2 | 82 | 10300 | 2.59 | 119.2 |
| P5 | 95.0: 5.0 | 95.4: 4.6 | 80 | 11200 | 2.52 | 119.1 |
The number- and weight-average molecular weights (Mn and Mw) of the copolymers determined by GPC are also shown in Table 1. The polydispersity indices (Mw/Mn) are similar and high (2.4–2.6).
T gs of the copolymers were determined by DSC and the DSC curves are shown in Fig. 2. All the copolymers were characterized by only one glass transition temperature (summarized in Table 1) indicating that they were most likely statistical, but not block copolymers. From Table 1, it could be found that the Tgs of the copolymers decreased gradually with the increase of the contents of MEDCA in the copolymers, probably due to the low Tg of PMEDCA (Tg = 117 °C).
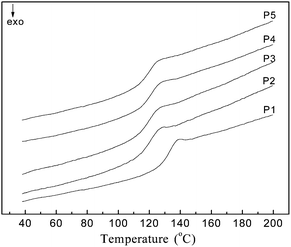 | ||
| Fig. 2 The DSC curves of the poly (NIPAM-co-MEDCA) obtained by a second heating process after quenching with heating rate of 10 °C min−1. The copolymers from P1 to P5 contain 1, 2, 3, 4, and 5 mol% MEDCA, respectively. | ||
Turbidimetry measurement
The LCSTs of all the copolymers were measured by cloud point (turbidity) measurements. The transmittance of UV-visible light was measured as a function of temperature (Fig. 3). At low temperature, the aqueous solution of copolymers was transparent and the transmittance of the light was high, while with increasing the temperature, the copolymer chains started to aggregate and phase separation occurred. The copolymer solution became cloud and the transmittance decreased rapidly as the solution became opaque. The transition should be attributed to the precipitation and/or sol to gel transformation of PNIPAM or related copolymers.24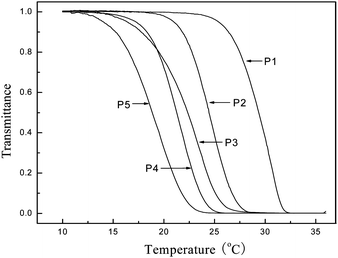 | ||
| Fig. 3 Light transmittance at 500 nm of the poly (PNIPAM-co-MEDCA) in 2.0 wt% aqueous solutions plotted as a function of temperature. The copolymers from P1 to P5 contain 1, 2, 3, 4, and 5 mol% MEDCA, respectively. | ||
According to LCSTs of copolymers summarized in Table 2, it could be found that the higher the content of MEDCA in the copolymer, the lower the resulting LCST of the copolymer. This can be explained tentatively as follows. There exists a local heterogeneity in the structure of water around NIPAM copolymer. Polar groups will form hydrogen bonds with water, whereas nonpolar sites are encapsulated in hydrophobic hydration shells. Below the LCST, hydrogen bonds between polar groups and water molecules energetically overcome the unfavorable decrease in entropy of the structured water in the hydrophobic shell and, therefore, the polymer is soluble in water. The hydrogen bonds are weakened with increasing temperature, and the situation is reversed above the LCST. Thus, the ratio of hydrophobicity/hydrophilicity of the copolymers as a whole are a main determinant of their LCST.4a The higher content of amphiphilic MEDCA moiety in the copolymers, the stronger hydrogen bonds between polar groups and water molecules as well as the stronger hydrophobicity of copolymers. Upon heating, the hydrogen bonds are weakened, which facilitated the corresponding copolymer with higher MEDCA content to form hydrophobic microdomain, causing a pronounced aggregation in solutions, thus resulting in easier phase separation at a lower temperature.
| Sample code | LCSTa °C | T gel b °C | T s c °C |
|---|---|---|---|
| a determined by turbidimetry. b visually determined when the polymer solutions did not flow by inverting the vials. c visually determined when the intact gel began to expel water slowly to form a shrunken mass by a vial tilt method. The copolymers from P1 to P5 contain 1, 2, 3, 4, and 5 mol % of MEDCA, respectively. | |||
| P1 | 29.4 | — | — |
| P2 | 24.5 | — | 29 |
| P3 | 22.3 | 27 | 31 |
| P4 | 21.3 | 26 | 33 |
| P5 | 18.8 | 24 | 34 |
Phase transitions
A typical phase transition process of an aqueous polymer solution (2 wt%) of P5 (i.e. 5 mol% MEDCA in feed) is shown in Fig. 4. | ||
| Fig. 4 Phase transitions and gelation process of an aqueous solution of the copolymer P5 (containing 5 mol% of MEDCA) with increasing temperatures. (Refer to the experimental section for the conditions of gelation process). The photos were taken at 2, 21, 28 and 40 °C from phase A to D, respectively. | ||
The clear polymer solution (Phase A) at the low temperature became cloud as temperature increases to its LCST of about 19 °C, but it was freely mobile (Phase B). With further increasing temperature, the cloud polymer solution subsequently became immobile at about 24 °C (defined as the gelation temperature, Tgel), resulting in gelation (Phase C) without a significant gel induction time. The time required for the gelation was apparently of the same order as the thermal induction time. The initially formed gel was translucent and became more opaque with increasing temperature. When the temperature increased to about 34 °C (defined as the gel-shrinking temperature, Ts), the gel started to shrink by expelling water, and finally formed a regular shrunken mass (Phase D). Once the gel was formed, it did not dissolve or swell as adding additional water. However, it became mobile upon cooling to the temperatures below that of their corresponding Tgel. It was notable, unlike the covalently crosslinked PNIPAM hydrogel, the shrunken gel dissolved directly without swelling when it was brought into an unstable temperature region suddenly.
Similarly, the phase transition behavior of aqueous solutions of other copolymers P1–P4 (i.e. 1, 2, 3, 4 mol% MEDCA in feed, respectively) was also investigated in detail. The phase transition behavior of all these copolymers is summarized in Fig. 5 as well as the corresponding Tgel and Ts are presented in Table 2. From Fig. 5 and Table 2, it could be seen that, similar to P5, P3 and P4 could form the intact gel at the temperatures of about 27 °C, 26 °C and subsequently formed regular shrunken gel at the elevated temperatures of about 31 °C, 33 °C, respectively. However, for P1, only the phase separation occurred and the gel could not be formed upon heating. In addition, the aqueous solution of P2 directly became a shrunken mass at elevated temperature without a gel phase. For P3–P5, it can be found there are interesting trends in the temperature ranges for each phase. With the increase of MEDCA content in the copolymers, the LCST decreases as presented in Fig. 3 and Table 2, the gelation temperature decreases, and the gel-shrinking temperature increases. These results suggest that the MEDCA content in the copolymers significantly influences their phase transition behavior.
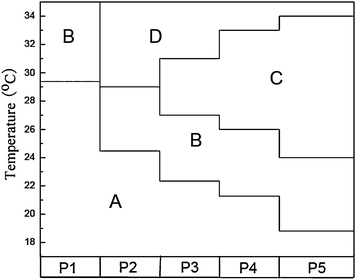 | ||
| Fig. 5 Phase transition temperatures of the copolymer solutions (2.0 wt% polymer). A: Clear solution phase; B: Cloud solution phase; C: Gel phase; D: Shrunken gel phase. The copolymers from P1 to P5 contain 1, 2, 3, 4, 5 mol% MEDCA, respectively. | ||
As we know, the phase separation of pure PNIPAM at a critical temperature is accompanied by a large structural change in the polymer chain, that is, a coiled chain collapses to a globular conformation. The conformational transition of PNIPAM from the coil to the globule is initiated by the dehydration of the polymer chain, which makes attractive interaction between hydrophobic isopropyl groups dominant and induces collapse of the chain.4 In the present work, the incorporation of only a few comonomer units containing amphiphilic deoxycholic acid ester into the PNIPAM chain obviously modified the phase transition behavior of the resultant copolymers according to the results above, most likely due to dominant control of amphiphilic deoxycholic acid ester in the balance between hydrophobicity and hydrophilicity of the copolymers.
Although the values for Ts may not be accurate, probably due to the occurance of spinodal decomposition,25 the illustration of the phase transition temperatures shown in Fig. 5 is still helpful for one to grasp the phase behaviors of the prepared hydrogels. There are some efforts made to understand the mechanism of the inverse thermally reversible gelation and the shrinkage of gel,19,26 however, it is still ambiguous to date. Thus, taking our observations into account, the phase transition behavior of all these copolymers can be tentatively explained as following.
Below the LCST, the predominant hydrogen bonding interaction between polar groups and water molecules leads to dissolution of the copolymer (Phase A). With increasing temperature, the hydrogen bonds are weaken, in turn, the hydrophobic interaction become predominant. The dehydration of the polymer chain with higher MEDCA content results in that a coiled chain collapses to a globular conformation, while the polymers with lower MEDCA content exists as a fully expanded coil state and/or as partially collapsed globules, which depends on the MEDCA content in the copolymers. The completely collapsed globules entangled with expanded coils may have a strong tendency to form hydrophobic aggregates with each other, resulting in a cloud solution (Phase B). The aggregates of collapsed chains form physical junctions by the main back-to-back interaction of the hydrophobic face of deoxycholic acid ester as well as the hydrogen bonding interaction of the hydroxyl groups within hydrophilic face of deoxycholic acid ester units,27 causing the formation of gel networks (Phase C). The physical gel formed is strong enough to inverse the vial. The strength of the formed gel increases with the increasing MEDCA content, probably due to that the higher MEDCA content, the higher the physical junction density of the gel networks. At elevated temperatures, the swollen gel starts to shrink by expelling water, which should be attributed to the further intrachain contraction, interchain association initiated by the further dehydration of the polymer chains with further increasing temperature, and finally resulting in a shrunken gel (Phase D). From Table 2, it can be found that Ts increased gradually as the increasing MEDCA content in the copolymer, which should be attributed to the higher stability of gel networks of the copolymers with higher MEDCA content because of the more physical junction density. The encapsulated water in gel networks of corresponding copolymer with higher MEDCA content is more difficult to lose than that of copolymer with lower MEDCA content.
According to the postulation above, the interesting result that P2 directly formed shrunken gel at elevated temperature (29 °C) without gel phase could also been explained as follows. Due to the lower content of MEDCA in the copolymer, resulting in the weaker interaction between deoxycholic acid ester groups, the copolymer P2 cannot form intact gel networks upon heating because of the lack of enough physical junctions. However, with further increasing the temperature, the hydrogen bonding interaction between polymer polar groups and water molecules is weakened, in turn, modifying the balance between hydrophilicity and hydrophobicity of the polymer chains, leading to the direct formation of shrunken gel.
Conclusions
In conclusion, a series of thermosensitive copolymers based on N-isopropylacrylamide and methacrylate derivatives of deoxycholic acid were successfully prepared by free radical polymerization. LCST behaviors of copolymers were determined with turbidimetry and the results indicated that the LCST of the copolymer was affected by the deoxycholic ester content, i.e., the higher content of MEDCA made the phase separation easier to occur at a lower temperature. This can be explained tentatively as follows. The hydrophilic face of deoxycholic acid ester can form hydrogen bonding with water, whereas its hydrophobic face has the tendency to form hydrophobic interaction. The latter is increased with increasing temperature resulting in a pronounced aggregation in solutions.An aqueous solution of the copolymer above a critical concentration (2 wt%) experiences four distinct phases of clear solutions, cloud solutions, gel and shrunken gel upon heating. Once the gel is formed, it does not dissolve or change water content until the gel-shrinking temperature arrives. The particular phase transition process is influenced by the MEDCA content in the copolymers and is related to molecular transition at elevated temperature from expanded coils to collapsed globules of a portion of the polymers, and the subsequent aggregation of the collapsed globules to make week physical junctions.
Acknowledgements
We would like to appreciate the National Natural Science Foundation of China (50703018, 20974055, 21074093) for financial support.Notes and references
- (a) F. Ganji and E. Vasheghani-Farahani, Iran. Polym. J., 2009, 18, 63 Search PubMed; (b) J. F. Mano, Adv. Eng. Mater., 2008, 10, 515 CrossRef CAS; (c) A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088 CrossRef CAS.
- (a) M. Tizzotti, M. P. Labeau, T. Hamaide, E. Drockenmuller, A. Charlot and E. Fleury, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2733 CrossRef CAS; (b) L. A. Lyon, Z. Y. Meng, N. Singh, C. D. Sorrell and A. S. John, Chem. Soc. Rev., 2009, 38, 865 RSC.
- F. Eeckman, A. J. Moës and K. Amighi, J. Controlled Release, 2003, 88, 105 CrossRef CAS.
- (a) Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2001, 17, 7535 CrossRef CAS; (b) B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37 CrossRef CAS.
- W. Xue, S. Champ and M. B. Huglin, Eur. Polym. J., 2001, 37, 869 Search PubMed.
- M. Shibayama, S. Y. Mizutzni and S. Nomura, Macromolecules, 1996, 29, 2019 CrossRef.
- M. S. Jones, Eur. Polym. J., 1999, 35, 795 Search PubMed.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352 CrossRef CAS.
- M. Kurisawa, M. Yokoyama and T. Okano, J. Controlled Release, 2000, 69, 127 CrossRef CAS.
- Y. Tsuda, A. Kikuchi, M. Yamato, Y. Sakurai, M. Umezu and T. Okano, J. Biomed. Mater. Res., 2004, 69A, 70 Search PubMed.
- S. Ibusuki, Y. Iwamoto and T. Matsuda, Tissue Eng., 2003, 9, 1133 Search PubMed.
- Y. Li and J. R. Dias, Chem. Rev., 1997, 97, 283 CrossRef CAS.
- A. Benrebouh, Y. H. Zhang and X. X. Zhu, Macromol. Rapid Commun., 2000, 21, 685 Search PubMed.
- S. Gouin, X. X. Zhu and S. Lehnert, Macromolecules, 2000, 33, 5379 CrossRef CAS.
- (a) J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241 CrossRef CAS; (b) I. S. Kim, Y. I. Jeong, C. S. Cho and S. H. Kim, Int. J. Pharm., 2000, 205, 165 CrossRef CAS.
- X. X. Zhu, D. Avoce, H. Y. Liu and A. Benrebouh, Macromol. Symp., 2004, 207, 187 Search PubMed.
- X. X. Zhu and M. Nichifor, Acc. Chem. Res., 2002, 35, 539 CrossRef CAS and references therein.
- (a) X. D. Wang, Y. Lu., Y. Q. Duan, L. H. Meng and C. X. Li, Adv. Mater., 2008, 20, 462 CrossRef CAS; (b) L. H. Meng, Y. Lu, X. D. Wang, J. Zhang, Y. Q. Duan and C. X. Li, Macromolecules, 2007, 40, 2981 CrossRef CAS; (c) J. Zhang, B. H. Zhao, L. H. Meng, H. Y. Wu, X. D. Wang and C. X. Li, J. Nanopart. Res., 2007, 9, 1167 CrossRef CAS; (d) J. Zhang, X. D. Wang, B. H. Zhao and C. X. Li, Chem. Lett., 2006, 35, 40 Search PubMed.
- C. K. Han and Y. H. Bae, Polymer, 1998, 39, 2809 CrossRef CAS.
- (a) N. J. Sarkar, J. Appl. Polym. Sci., 1979, 24, 1073 CrossRef CAS; (b) T. Kato, M. Yokoyama and A. Takahashi, Colloid Polym. Sci., 1978, 256, 15 CAS.
- R. A. Barabshina and O. M. Sleptsava, Zh. Prikl. Khim., 1967, 40, 696 Search PubMed.
- A. Benrebouh, D. Avoce and X. X. Zhu, Polymer, 2001, 42, 4031 Search PubMed.
- H. Y. Liu, D. Avoce, Z. J. Song and X. X. Zhu, Macromol. Rapid Commun., 2001, 22, 675 Search PubMed.
- (a) D. Avoce, H. Y. Liu and X. X. Zhu, Polymer, 2003, 44, 1081 Search PubMed; (b) C. Y. Choi, S. Y. Chae and J. W. Nah, Polymer, 2006, 47, 4571 Search PubMed; (c) R. Salehi, N. Arsalani, S. Davaran and A. A. Entezami, J. Biomed. Mater. Res., Part A, 2009, 89A, 919 Search PubMed.
- A. Onuki and S. Puri, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 59, R1331 Search PubMed.
- J. Zhou, G. Huang, M. Li and A. K. Soh, Modell. Simul. Mater. Sci. Eng., 2010, 18, 025002 Search PubMed.
- (a) J. Zakrzewska, V. Markovic, D. Vucelic, L. Feigin, A. Dembo and L. Mogilevsky, J. Phys. Chem., 1990, 94, 5078 CrossRef CAS; (b) P. Venkatesan, Y. Cheng and D. Kahne, J. Am. Chem. Soc., 1994, 116, 6955 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |