Single site metallorganic polymerization catalysis as a method to probe the properties of polyolefins
Claudio
De Rosa
* and
Finizia
Auriemma
Dipartimento di Chimica “Paolo Corradini, Università di Napoli “Federico II”, Complesso Monte S.Angelo, Via Cintia, I-80126, Napoli, Italy. Tel: +39 081 674341; E-mail: claudio.derosa@unina.it; auriemma@unina.it; Fax: +39 081 674090; Tel: +39 081 674346
First published on 9th June 2011
Abstract
Single-site metallorganic catalysts for the polymerization of olefins has allowed the synthesis of new polymeric materials having molecular structures that cannot be obtained with conventional Ziegler–Natta catalysts, with exceptional control over the chain architecture, including control of the concentration of stereo- and regio-defects and efficient uniform placement of comonomeric units along the chains. The cases of isotactic and syndiotactic polypropylenes are illustrated as examples of a fine tuning of the physical and mechanical properties of polymeric materials achieved through the controlled incorporation of stereo- and regio-defects, and the rational choice of the catalytic system. Novel isotactic polypropylenes with controlled properties, intermediate between those of stiff-plastic and elastomeric materials have been produced through the design of the catalysts. For syndiotactic polypropylene, a new class of thermoplastic elastomers with finely controlled mechanical properties have been produced by using metallorganic catalysts of different stereoselectivity. Depending on the concentration of stereodefects, elastomers showing entropic or unconventional enthalpic elasticity, can be produced.
 Claudio De Rosa | Claudio De Rosa is Full Professor of Macromolecular Chemistry at the University of Naples “Federico II” (Italy). He received his Doctor degree in Chemistry in 1983 (Magna Cum Laude) and the Ph.D. in Chemistry in 1989 from the University of Naples. He was assistant professor of Inorganic Chemistry at the University of Naples from 1990 to 1992 and Associate Professor of Industrial Chemistry from 1992 to 2002. He has been Full Professor since 2002 and head of the Chemistry Department of the University of Naples since 2008. His research activity comprises the study of the crystal structure of polymers and the interplay between physical properties and the molecular structure of polymers. He is author of about 220 papers published in international scientific journals, various patents and chapters of books. |
 Finizia Auriemma | Finizia Auriemma is Associate Professor of Macromolecular Chemistry at the University of Naples “Federico II” (Italy). She received her Doctor degree in Chemistry in 1984 (Magna Cum Laude) and the Ph.D. in Chemistry in 1989 from the University of Naples. She was assistant professor of Inorganic Chemistry at the University of Naples from 1990 to 2002 and has been Associate Professor of Industrial Chemistry since 2002. Her research activities comprise the theoretical and experimental studies of the crystal structure of polymers and of the disorder in polymer crystals, aimed at finding relationships between physical and mechanical properties and the molecular and the crystalline structures of polymers. She is author of more than 150 papers published in international scientific journals and various patents and chapters of books. |
1 Introduction
The development of single-site metallocene catalysts for the polymerization of olefins has allowed production of new materials having molecular structures (chain microstructures) that cannot be obtained with conventional Ziegler–Natta catalysts.1–15 These catalysts afford exceptional control over chain architecture, including control of concentration of stereo- and regio-defects and efficient uniform placement of constitutional defects, as comonomeric units, along the chains,1–15 with relevant impact on the final material properties. A major evolution has been achieved in the field of polypropylene, which can now be produced with any type and degree of stereoregularity,5–9 from highly isotactic to highly syndiotactic (Fig. 1).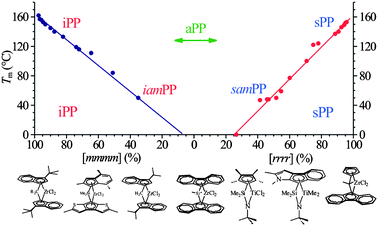 | ||
| Fig. 1 Melting temperature of polypropylene as a function of stereoregularity expressed as concentration of isotactic mmmm and syndiotactic rrrr pentads. Depending on the structure of the catalyst, highly isotactic (iPP), poorly isotactic, nearly amorphous (iamPP) polypropylene, fully atactic polypropylene (aPP), highly syndiotactic (sPP) and poorly syndiotactic-nearly amorphous (samPP) polypropylene, can be produced.16,17,19,21,25,26 | ||
In particular, in the case of isotactic polypropylene (iPP), a fine tuning of the crystallization and physical properties is possible by tailoring the chain microstructure, through the controlled incorporation of stereo- and regio-defects16–20 and constitutional defects,20 which, in turn, can be achieved through the rational choice of the metallorganic complex precursor of the catalytic system.16–20 Polypropylenes characterized by different kinds and amounts of regio- and stereo-irregularities, different distributions of defects and different molecular masses, and physical properties that range between those of stiff materials and elastomers, can be obtained.16–20 The achieved profound insight into the relationships between catalyst structure, chain microstructure, crystallization properties of iPP and final product properties has allowed tailoring of polymer properties in the polymerization stage.
In this review the cases of isotactic16–20 and syndiotactic21–28 polypropylenes prepared with metallocene catalysts are illustrated as examples of a fine tuning of the physical and mechanical properties of polymeric materials achieved through the controlled incorporation of stereo- and regio-defects and constitutional defects, and the rational choice of the catalytic system. As discussed above and shown in Fig. 1, depending on the structure of the catalyst, polypropylenes (PP) of virtually any microstructure from fully atactic to highly isotactic and syndiotactic PP can be obtained, with gradual change of the melting temperature. Along the straight lines of decrease of melting temperature of Fig. 1 a dramatic change of the mechanical behavior is also observed.16,17,19,21,25,26 These features are unique and are not shown by polypropylene prepared with conventional Ziegler–Natta catalysts. For this reason the wide range of polypropylenes of different properties prepared with metallocene catalysts are generally indicated as metallocene-made polypropylenes.
The example of polypropylene shows that the use of these unique methods of controlled synthesis has allowed a full understanding of the relationships between catalyst structure, chain microstructure, crystallization properties and final material properties. The mechanical properties of these materials are largely related to the crystal structure and morphology, which in turn depend on the chain microstructure generated by the specific catalyst used.16–28 Moreover, the possible occurrence of polymorphic transformations during plastic deformation induced, for instance, by application of uniaxial stretching, plays a fundamental role and may explain unexpected flexibility and ductility, even in the case of highly crystalline samples.19,21,28
2 Isotactic polypropylene
The wide application range of iPP results from its versatility and the variety of possible modifications of the basic material, which already starts in the polymerization reactor. The main structural factors affecting the physical properties are stereoregularity, molecular mass, and polydispersity, mostly through their influence on crystallinity. Tailoring polymer properties in the polymerization stage has required a profound insight into the relationships between catalyst structure, chain microstructure, crystallization properties, and final product properties.16–20,29–41As discussed above, the development of single-site metallocene catalysts has allowed synthesizing polypropylenes with controlled chain microstructure. Polypropylenes characterized by different kinds and amounts of regio- and stereo-irregularities, different distributions of defects, and different molecular weights, can be prepared through the rational choice of the catalytic system.5–9 The possibility to synthesize samples of iPP containing a controlled concentration of a single type of microstructural defect, for instance only isolated defects of stereoregularity generating a rr triad,16 or only defects of regioregularity due to secondary 2,1 insertions of monomer in a prevailing primary 1,2 enchainment,18 has afforded the opportunity to study the effect of a single type of defect on the crystallization behavior and mechanical properties of iPP.16–20,29–35 Moreover, in chains of iPP samples prepared with single-site metallocene catalysts the distribution of defects is random and the length of fully isotactic sequences is roughly inversely related to the content of defects. In these model systems the crystallization behavior and the physical properties are directly related to the type and concentration of incorporated defects. The full understanding of the structure-properties relationships has contributed to further increase the control over the physical properties of polypropylene and possibly further expand its application range.
2.1 Influence of stereodefects on the crystallization behavior
A unified view of the crystallization behavior of metallocene-made polypropylene has been recently suggested on the basis of the structural study of a series of iPP samples having a simple and controlled molecular structure, characterized by the presence of a single type of defect.16–19,29–35Stereodefective and regioregular samples of iPP containing only rr triad stereodefects in a wide range of concentration and free from regioerrors have been prepared with highly regiospecific catalysts of Fig. 2,8,9 activated with methylaluminoxane. These catalysts produce primary insertions of the monomer with the wrong enantioface, generating isolated rr triads, whose concentration depends on the structure of the zirconocene complex, in particular the indenyl substituents, and can be varied in the range between 0.5 and 17%.16,17,19,29 Correspondingly the samples show melting temperatures variable between 162 and 45 °C.16,17,19,29 The produced samples provide a model system of iPP with a random distribution of defects, containing only one type of stereodefect with a concentration spanning a wide range and not including regio-errors.
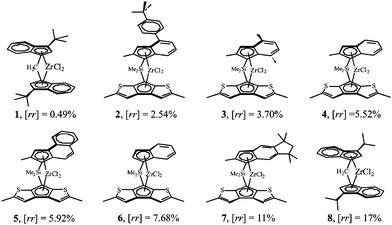 | ||
| Fig. 2 Structures of the zirconocene complexes used as precursors of the catalysts for the polymerization of propene and the synthesis of streodefective isotactic polypropylene. The concentration of the rr triad stereodefects produced by each catalyst are indicated.5–9 | ||
The X-ray powder diffraction profiles of samples crystallized by slow cooling from the melt by compression-molding (Fig. 3) indicate that these samples crystallize from the melt as mixtures of α and γ forms (Fig. 4A and B, respectively),16,17 as indicated by the presence of both (130)α and (117)γ reflections of α and γ forms, respectively, in the diffraction profiles of Fig. 3a–f. The most isotactic sample crystallizes basically in the α form (Fig. 3a), with a limit low concentration of γ form of 15–20%, as indicated by the high intensity of the (130)α reflection at 2θ = 18.6° and the low intensity of the (117)γ reflection of γ form at 2θ = 20.1°. The intensity of the (117)γ reflection of the γ form at 2θ = 20.1°, increases with increasing concentration of rr defects, indicating that the relative amount of crystals of γ form increases with increasing concentration of rr defects, up to 100% for rr concentrations higher than 6–7% (Fig. 5A).16,17,19,29 For samples isothermally crystallized from the melt16 the fraction of γ form increases with increasing crystallization temperature and the concentration of rr defects16,33–39 and a linear relationship between the maximum amount of γ form and the average length of isotactic sequences has been found.16
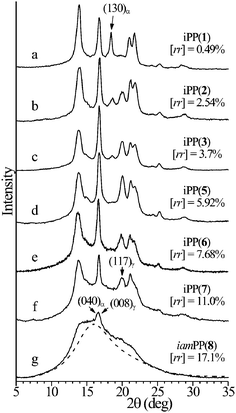 | ||
| Fig. 3 X-ray powder diffraction profiles of iPP samples containing the indicated concentration of rr triad stereodefects and prepared with the catalysts in Fig. 2, crystallized from the melt by compression molding and cooling the melt to room temperature at 1 °C min−1.17,19,29 The dashed line indicates the diffraction profile of the amorphous phase. The (130)α reflection of the α form at 2θ = 18.6° and the (117)γ reflections of the γ form at 2θ = 20.1° are indicated. The (040)α and (008)γ reflections at 2θ = 17° of α and γ forms, respectively, are also indicated.17,19 The numbers in parenthesis indicate the catalysts of Fig. 2. | ||
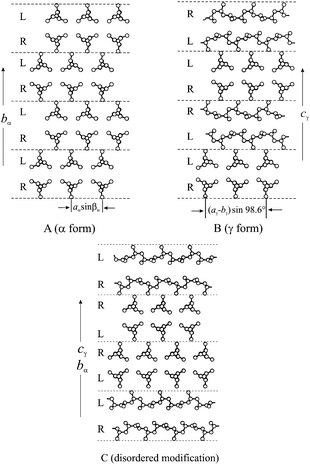 | ||
| Fig. 4 Limit ordered models of packing proposed for α (A) and γ (B) forms of iPP and model of the α/γ disordered modifications intermediate between α and γ forms (C). In the disordered model (C) consecutive bilayers of chains are stacked along bα (cγ) with the chain axes either parallel or nearly perpendicular, making α-like or γ-like arrangements of bilayers.32 | ||
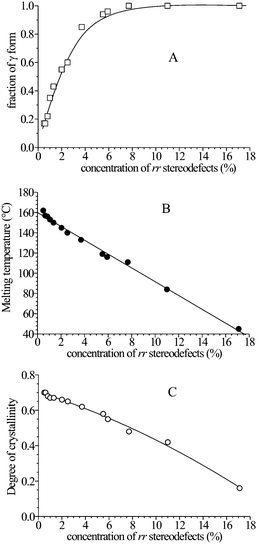 | ||
| Fig. 5 Relative amount of γ form (A), melting temperature (B) and degree of crystallinity (C) in the stereodefective iPP samples crystallized from the melt by compression moulding and cooling the melt to room temperature at cooling rate of 1 °C min−1, as a function the concentration of rr defects.17,19 | ||
The presence of rr-defects, therefore, induces crystallization of the γ form. Crystals of the γ form obtained in these samples always present structural disorder characterized by defects in the regular packing of bilayers of chains with axes oriented alternatively along two nearly perpendicular directions.16,17,19,29–34 This disorder produces a local situation of packing typical of the α form, with some adjacent bilayers having parallel chains (Fig. 4C, α/γ disorder). Therefore metallocene-made iPP crystallizes in a continuum of disordered modifications intermediate between α and γ forms (Fig. 4), the amount of disorder being dependent on the crystallization conditions and on the stereoregularity of the sample.32,34
The iPP sample of lowest stereoregularity of Fig. 3g (sample iamPP), prepared with the catalyst 8 of Fig. 2, does not crystallize by cooling the melt to room temperature, but slowly crystallizes in disordered modifications intermediate between α and γ forms (Fig. 4C) if the sample cooled from the melt is kept at room temperature for several days.29 In fact, the X-ray diffraction profile of Fig. 3g presents only a sharp reflection at 2θ = 17°, corresponding to the (040)α reflection of α form or the (008)γ reflection of γ form. The other sharp Bragg reflections of both α and γ forms, as (110)α and (130)α reflections at 2θ = 14° and 18.6°, respectively, typical of the α form, and (111)γ and (117)γ reflections at 2θ = 14 and 20.1°, respectively, typical of the γ form are absent. This indicates that the sample iamPP does not crystallize in the pure α or γ forms, but in a disordered modification intermediate between α and γ forms (Fig. 4C).29,32 Although this sample is a nearly atactic polypropylene with a prevailingly isotactic microstructure ([mmmm] ≈ 35%) and a high concentration of randomly distributed stereodefects ([rr] ≈ 17%), it is still able to crystallize,29 suggesting a particular role for rr stereodefects, even at high concentrations, on the crystallization of iPP.
The melting temperature and the degree of crystallinity of samples crystallized from the melt of Fig. 3 gradually decrease with increasing the content of rr defects (Fig. 5). The degree of crystallinity (Fig. 5C) decreases only slightly with increasing concentration of rr defects in the range 0–11%, then drops to very low values (≈16%) for the less stereoregular sample iamPP.29
The possible crystallization of iPP in disordered modifications of the type of Fig. 4C, intermediate between α and γ forms and containing high degree of α/γ disorder, and the inclusion of rr defects in these defective and disordered α/γ crystals29,32 allows maintenance of crystallinity even in poorly isotactic, basically atactic, samples, containing high concentration of rr defects, as the sample iamPP containing 17% of rr defects.16,29,32 The presence of this low level of crystallinity (≈16%) determines the outstanding properties of the poorly isotactic sample iamPP,29 that shows elastic properties, high flexibility, transparency and is non-sticky.29 Moreover these poorly isotactic, almost amorphous polypropylenes (iamPP) show better miscibility with iPP compared to fully atactic polypropylene, leading to flexible polypropylene blends with improved transparency.
2.2 Influence of stereo-defects on the mechanical properties
The stress-strain curves of compression-molded films of some stereodefective iPP samples are reported in Fig. 6.16,17,19 The values of Young modulus decrease with increasing concentration of rr defects and decreasing crystallinity, from nearly 200 MPa of the more isotactic sample17 to nearly 1 MPa of the poorly stereoregular sample iamPP (Fig. 7A).29 The values of deformation at break, instead, increase with increasing concentration of defects (Fig. 7B). Parallel to the decrease of the values of the modulus, a decrease of the stress at yielding with increasing concentration of defects is observed.17,19,29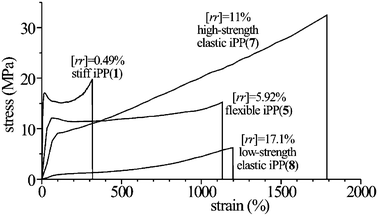 | ||
| Fig. 6 Stress-strain curves of iPP samples of different stereoregularity containing only rr triads defects of stereoregularity prepared with the catalysts of Fig. 2. The values of the concentration of the rr stereodefects is indicated.16,17,19 The numbers in parentheses indicate the catalysts of Fig. 2 used for the synthesis of the various samples. (Reproduced with permission from ref. 19. Copyright 2006 by the American Chemical Society). | ||
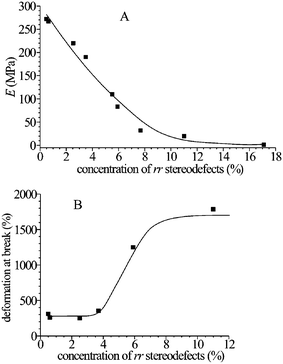 | ||
| Fig. 7 Values of elastic modulus E (A) and deformation at break (B) as a function of concentration of rr defects of iPP samples prepared with the catalysts of Fig. 2.16,17,19,20 | ||
Samples of iPP containing low concentration of rr defects (up to [rr] = 3.7%, Fig. 6) show non uniform stretching behavior and high values of the elastic modulus (Fig. 7) typical of stiff-plastic materials. Nevertheless, they present high ductility. In fact, these samples can be stretched at room temperature up to remarkable values of the strain (250–350%).17 We recall that highly stereoregular and crystalline iPP samples, prepared with Ziegler–Natta catalysts, conventionally crystallized from the melt and showing the typical cross-hatched α form lamellar morphology and spherulitic superstructure, present a rather brittle behavior with values of strain at break of the order of only 10%. A post-yield deformation region is generally not detected for highly crystalline samples, that is, plastic crystal deformation and fibrillation is not observed. Highly isotactic samples of iPP prepared with the metallocene catalyst 1, containing low concentration of rr defects ([rr] = 0.49%), show, instead, good ductility, with values of strain at break of 200–300% (Fig. 7).17
A further strong increase of the ductility and toughness is observed for higher contents of rr defects (Fig. 6). In particular, samples with concentration of rr defects around 5–6% still present behavior of thermoplastic materials with slightly lower strength but much higher deformation at break (εb ≈ 1200%). This indicates that these samples ([rr] = 5–6% and melting temperatures around 110–115 °C) behave as highly flexible thermoplastic materials.17,19
The less crystalline samples, with concentration of rr defects in the range 7–11% show strong strain hardening at high deformation (Fig. 6) with values of the tensile strength (32 MPa) higher than those of the more isotactic and crystalline samples (20–23 MPa).17,19 The strain hardening may be somehow related to the fact that these samples present crystals with low plastic resistance and uniform stretching behavior. Once the crystals have experienced irreversible plastic deformations at low values of deformation, strain hardening is caused by straightening of the entangled network. Similar strain hardening is experienced also by the lowest isotactic and crystalline sample iamPP,29 even though the achieved values of the tensile strength and the values of the stress at any strain are one order of magnitude lower than those of samples wit [rr] = 7–11%, because of the much lower level of crystallinity.
Samples with the highest concentration of rr defects ([rr] = 7–17%) show elastomeric properties.16,17,19,29 The stress-strain curves of these samples present, indeed, typical shape of elastomeric materials (Fig. 6), showing high values of deformation at break and strain-hardening at high deformations. In the samples with [rr] = 7−11%, the elastic properties are associated with remarkable values of the modulus of nearly 20–30 MPa (Fig. 6 and 7A),16,17 and, as discussed before, very high values of tensile strength.17 In the case of the poorly stereoregular sample iamPP, low values of the tension set are observed even at high deformation, indicating that the sample iamPP experiences a recovery of the initial dimension after breaking as well as after removing the tension from any deformation.29
The elastic behavior of samples with [rr] = 7−17% is due to the fact that these samples are crystalline notwithstanding the low stereoregularity. These samples, indeed crystallize in the γ form of iPP or in α/γ disordered intermediate modifications thanks to the inclusion of most of the rr stereo-defects (profiles e–g in Fig. 3).16,17,19,29 The formation of small crystalline domains induces elastomeric properties since crystals act as physical cross-links in the amorphous matrix, producing the elastomeric network. The presence of a high level of crystallinity (40–45%, Fig. 5C) of the samples with defect concentration up to [rr] = 11%, gives high values of the strength, so that interesting thermoplastic elastomers with remarkable values of the modulus and tensile strength are obtained.16,17,19 The small degree of crystallinity of the poorly stereoregular sample iamPP (≈16%), induces good elastic properties in a range of deformation larger than that shown by more isotactic samples.29 The small crystalline domains of highly disordered α/γ modifications that develops upon aging of the amorphous sample obtained by cooling the melt to room temperature, act as physical knots of the elastomeric network, preventing the viscous flow of the amorphous chains and giving a typical thermoplastic elastomeric behavior.29 The poorly isotactic sample iamPP shows, indeed, poor elastic properties and viscous flow at high deformations in the amorphous state, before crystallization.29
It has been argued that the outstanding mechanical properties of metallocene-made iPP samples containing only rr defects, is related to the easy inclusion of rr defects inside the crystalline phase.16–19 The high ductility and good drawability at room temperature of these materials even when the concentration of rr defects is low and the samples basically crystallize in the α form, may be indeed explained by the fact that rr stereodefects are uniformly distributed between crystalline and amorphous phases, which have, therefore, the same composition. First of all the presence of defects in the crystals of these materials reduces their plastic resistance. Moreover, the similar composition of the crystalline and amorphous phases makes the process of plastic deformation easier, reducing the stress level necessary for the destruction of the preexisting lamellae and re-crystallization of chains in new oriented crystallites with fibrillar morphology.
The continuous change of mechanical properties of metallocene-made iPP samples as a function of concentration of rr defects of stereoregularity is shown in Fig. 8.16,17,19 Samples with low concentration of rr defects, up to 3–4%, present high melting temperatures, in the range 162–130 °C, and behave as stiff plastic materials, with high values of Young modulus.16,17,19 Sample with higher rr content, in the range 4–6%, and melting temperatures around 115–120 °C are highly flexible thermoplastic materials, showing very high deformation at break. Samples with concentration of rr defects in the range 7–11% and melting temperature in the range 80–110 °C are thermoplastic elastomers with high strength.16,17,19 Samples with concentration of rr defects higher than 11% and melting temperature lower than 50 °C are soft materials with elastomeric properties more similar to those of conventional thermoplastic elastomers.29
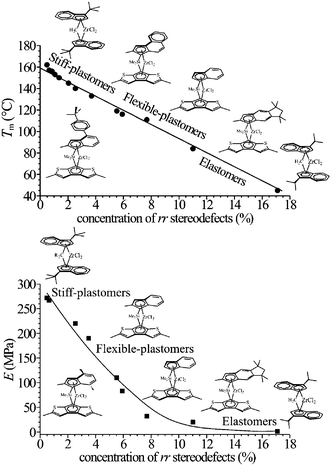 | ||
| Fig. 8 Classification of iPP samples prepared with different metallocene catalysts of Fig. 2, as stiff-plastic materials, flexible-plastic materials, and thermoplastic elastomers depending on the concentration of rr defects of stereoregularity and values of melting temperature (top) and Young modulus (bottom) as a function of rr concentration.16,17,19,20 | ||
2.4 Stress-induced phase transformations of iPP during tensile deformation
The different mechanical behavior of stereodefective iPPs is also related to the structural transformations that occur during stretching.16,17,19 The more isotactic samples, initially crystallized in the α form (Fig. 3a) partially transform by stretching into the mesomorphic form of iPP already at low deformation (Fig. 9A). The formation of the mesomorphic form is indicated by the presence of a broad halo in the range of 2θ = 14–18° (Fig. 9A). A fraction of crystals of α form is still present even at the maximum possible deformation (Fig. 9B). These crystals have high plastic resistance and do not allow further deformation. Samples with low concentration of rr defects, up to 3–4%, behave as stiff materials, but they can be easily stretched up to 200–300% strain, because the fraction of crystals that undergoes plastic deformation without sample breaking transforms into the mesomorphic form that, in turn, facilitates successive deformation (Fig. 6). | ||
| Fig. 9 X-ray fiber diffraction patterns of fibers of iPP samples of different stereoregularity stretched at the indicated values of deformation ε.17,19 (Reproduced with permission from ref. 19. Copyright 2006 by the American Chemical Society). | ||
For more flexible samples with rr contents in the range 4–11% the γ form present in compression-molded films (Fig. 3b–f) gradually transforms into the mesomorphic form by stretching (Fig. 9C,D). In these samples, the low crystallinity and the presence of high degrees of structural disorder16,17 reduce the stress level necessary for inducing plastic deformation of crystals. The formation of the mesomorphic form after yielding, which is consequent to the easy plastic deformation, facilitates successive further deformation up to very high strains (Fig. 6). The presence of rr stereodefects included in crystals of γ form and the formation of the mesophase produce the development of outstanding properties of high flexibility and elasticity, which are unusual for polypropylene.17,19
The nearly atactic and amorphous sample iamPP with high concentration of rr defects (17 mol %) slowly crystallizes upon aging at room temperature or by stretching in a disordered modification intermediate between α and γ forms (Fig. 3g), with a maximum degree of crystallinity of only 16% (Fig. 5C).29,32 The stretching of this sample, even at high deformation, does not produce formation of the mesomorphic form, probably due to the breaking of the sample prior to the transition, but only α/γ disordered modifications, more similar to the α form (Fig. 4C), are obtained (Fig. 9E,F).29
These data have allowed building the phase diagram of iPP at room temperature (Fig. 10), where the regions of stability of the different polymorphic forms of iPP in oriented fibers are defined as a function of stereoregularity and degree of deformation.19 The values of the critical strain at which the polymorphic transitions start, and at which the transformation is complete, depend on the stereoregularity.19
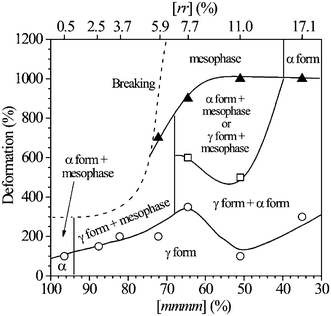 | ||
| Fig. 10 Phase diagram of iPP showing the region of stability of the different polymorphic forms as a function of deformation ε (ε = 100(Lf − L0)/L0) and stereoregularity, defined as concentration of the fully isotactic pentads mmmm. The values of critical strains corresponding to the boundary lines between the various crystalline forms have been determined from the X-ray fiber diffraction patterns of iPP samples of different stereoregularity prepared with the catalysts of Fig. 2 (as those of Fig. 9). The concentration of rr triad defect is indicated in the upper scale. The deformation at break is also indicated as a dashed line.19 (Reproduced with permission from ref. 19. Copyright 2006 by the American Chemical Society). | ||
Inspection of Fig. 10 suggests that large values of ductility may be achieved thanks to the occurrence of phase transitions. During stretching, the mechanical energy is indeed converted into a latent heat of fusion that induces local melting of crystals, followed by recrystallization into a new form and, at high deformation, into the mesophase. The transformation of α or γ forms into the mesomorphic form plays the most important role in defining the mechanical behavior.19
The phase diagram of Fig. 10 provides an easy picture of the conditions of formation of the mesomorphic form, in terms of microstructure and processing, which is the basis for the development of the unusual mechanical properties of metallocene-made iPP.19
The results of Fig. 6–10 demonstrate the novel utility of single-site metallocene catalysts in that precise correlation between the chemical structure of the catalyst precursor, the chain microstructure, crystallization behavior, polymorphic transitions, and mechanical properties may be established. A detailed understanding of the relationships between the molecular structure and the properties of these materials, may afford the tailored design of new polymerization catalysts for producing polypropylenes having controlled microstructures and controlled desired properties, intermediate between those of stiff-plastic and elastomeric materials, not accessible to commercial polypropylene produced with traditional Ziegler–Natta catalysts.16–20
2.5 Elastic properties and phase transformations in stereodefective iPP
As discussed above, the stereodefective iPP samples with concentration of rr defects higher than 7–8 mol% are thermoplastic elastomers (Fig. 6). In particular, the sample with [rr] = 11% prepared with the catalyst 7 of Fig. 2, shows elastic properties with remarkable values of modulus and strength and non negligible level of crystallinity (40–45%),16 whereas the most stereoirregular sample iamPP with [rr] = 17%, prepared with the catalyst 8 of Fig. 2, shows elastic properties with low modulus and strength, due to the lower value of crystallinity (≈16%) (Fig. 6).29Oriented fibers of these samples show a perfect elastic behavior after stretching and relaxation, as shown by the stress-strain hysteresis curves recorded in successive cycles of stretching and relaxation reported in Fig. 11.16,17,29 It is apparent that successive cycles of stretching and relaxation, measured after the first one, are in all cases nearly coincident, indicating a tension set (the residual deformation after relaxation) close to zero and a perfect elastic recovery.17,29
![Stress-strain hysteresis cycles recorded at room temperature, composed of stretching and relaxation (at controlled rate) steps according to the direction of the arrows, for stress-relaxed fibers of stereoirregular samples of iPP with [rr] = 11%16,17,19 (A) and [rr] = 17.1% (sample iamPP)29 (B). The stress-relaxed fibers have been prepared by stretching compression-molded films, of initial length L0, up to 1000% (A) or 600% (B) elongation (final lengths Lf = 11L0 or 7L0, respectively), and, then, removing the tension. In the hysteresis cycles the stretching steps are performed stretching the fibers up to final length Lf = 11L0 for the sample in A and Lf = 7L0 for the sample iamPP in B. (A) Continuous lines: first cycle; dashed lines: second cycle; dotted lines: third cycle and successive cycles. (B) Continuous lines: first cycle; dashed lines: curves averaged for at least four cycles successive to the first one. (Reproduced with permission from ref. 17, copyright 2005 by Elsevier (A) and from ref. 29, copyright 2004 by the American Chemical Society (B).](/image/article/2011/PY/c1py00129a/c1py00129a-f11.gif) | ||
| Fig. 11 Stress-strain hysteresis cycles recorded at room temperature, composed of stretching and relaxation (at controlled rate) steps according to the direction of the arrows, for stress-relaxed fibers of stereoirregular samples of iPP with [rr] = 11%16,17,19 (A) and [rr] = 17.1% (sample iamPP)29 (B). The stress-relaxed fibers have been prepared by stretching compression-molded films, of initial length L0, up to 1000% (A) or 600% (B) elongation (final lengths Lf = 11L0 or 7L0, respectively), and, then, removing the tension. In the hysteresis cycles the stretching steps are performed stretching the fibers up to final length Lf = 11L0 for the sample in A and Lf = 7L0 for the sample iamPP in B. (A) Continuous lines: first cycle; dashed lines: second cycle; dotted lines: third cycle and successive cycles. (B) Continuous lines: first cycle; dashed lines: curves averaged for at least four cycles successive to the first one. (Reproduced with permission from ref. 17, copyright 2005 by Elsevier (A) and from ref. 29, copyright 2004 by the American Chemical Society (B). | ||
The elastic recovery observed in the stress-strain curves of Fig. 11 is associated with reversible polymorphic transformations, which occurs during stretching and relaxation. X-ray diffraction patterns recorded during stretching of the samples iPP(7) and iamPP(8) with [rr] = 11 and 17%, respectively, are reported in Fig. 12. Fibers of the sample with [rr] = 11% stretched at high deformation are in the pure mesomphic form (Fig. 9D and 12A). Crystals of the γ form of the unoriented compression-molded film transform by stretching into the mesophase at high deformations (Fig. 9C,D). The mesomorphic form obtained by stretching, transforms into the α form by releasing the tension (Fig. 12A,B).16,17,19 This transformation is reversible upon successive stretching and relaxing cycles and is associated with the fully elastic recovery of the sample (Fig. 11A and 12A,B). The crystalline α form transforms again by stretching into the disordered mesomorphic form, which, in turn, transforms back into the α form by releasing the tension (Fig. 12A,B).16,17
![X-ray fiber diffraction patterns of fibers of the stereodefective samples iPP(7) with [rr] = 11% (A,B) and iamPP(8) with [rr] = 17% (C,D), obtained by stretching compression molded films at 1000% elongation, keeping the fiber under tension (A,C) and after removing the tension (B,D).16,17,19,29 (A,B: Reproduced with permission from ref. 16. Copyright 2004 by the American Chemical Society. C,D: Reproduced with permission from ref. 29. Copyright 2004 by the American Chemical Society).](/image/article/2011/PY/c1py00129a/c1py00129a-f12.gif) | ||
| Fig. 12 X-ray fiber diffraction patterns of fibers of the stereodefective samples iPP(7) with [rr] = 11% (A,B) and iamPP(8) with [rr] = 17% (C,D), obtained by stretching compression molded films at 1000% elongation, keeping the fiber under tension (A,C) and after removing the tension (B,D).16,17,19,29 (A,B: Reproduced with permission from ref. 16. Copyright 2004 by the American Chemical Society. C,D: Reproduced with permission from ref. 29. Copyright 2004 by the American Chemical Society). | ||
It is worth noting that the transformation of the disordered mesomorphic form into the α form corresponds to an increase of crystalline order. The crystallization upon removing the tension in stretched fibers is not common in polymers and is opposite to what is generally observed in a common elastomer as the natural rubber,42 for which crystallization occurs during stretching, whereas melting occurs upon releasing the tension. The crystallization of the mesomorphic form into the α form upon releasing the tension is not observed in the case of flexible or stiff-plastic samples having rr concentration lower than 7 mol%, which do not show elastic behavior. In these sample the mesomorphic form obtained by stretching at high deformations is stable after removing the tension and does not transform into the α form.17 This indicates that in the elastomeric iPP samples elasticity is probably partially due to the enthalpic contribution associated with the crystallization of the mesomorphic form into the α form upon relaxation.16,17,19
Also in the case of the poorly isotactic sample iamPP the elastic recovery observed upon releasing the tension (Fig. 11B) is associated with a polymorphic transformation occurring inside the crystalline domains. As shown in Fig. 9F and 12C, oriented fibers of the sample iamPP are in α/γ disordered modifications very close to the α form, having a high fraction of consecutive bilayers facing as in the α form with parallel chain axes. These disordered modifications more similar to the α form obtained by stretching transform back into more disordered modifications closer to the γ form upon releasing the tension (Fig. 12D), with a simultaneous decrease of the degree of orientation of the crystals.29 This transformation is reversible upon successive stretching and relaxing cycles and is associated with the fully elastic recovery of the sample (Fig. 11B and 12C,D).29
The crystallization of this sample upon aging and stretching and the formation of the continuum of α/γ disordered modifications, due to the inclusion of the rr stereodefects in the disordered crystals, induces good elastic properties in a large range of deformation allowing the formation of the elastomeric network. The small crystals that develop upon aging act as knots of the lattice preventing viscous flow of the chains and play an active role thanks to the reversible phase transformation occurring during stretching and relaxation.29
3 Syndiotactic polypropylene
The discovery of the single-site metallocene catalysts also allowed the synthesis of highly stereoregular and regioregular syndiotactic polypropylene (sPP), which could not be obtained with Ziegler–Natta catalysts.1,2,21 The improved stereoregularity and molecular mass has allowed finding outstanding and unique mechanical properties, the most important one relies on the elastomeric behavior, notwithstanding the high crystallinity and the relatively high glass transition temperature.21–28The physical properties of sPP basically depend on the crystallization behavior, which, in turn, is strongly related to the chemical structure of the single macromolecules.21 Depending on the catalyst and condition of polymerization it is nowadays possible to tailor tacticity and melting temperature and different syndiotactic polypropylene samples, from only slightly syndiotactic fully amorphous to highly syndiotactic and crystalline samples, having different physical properties can be produced.21
Some important catalysts that produce sPP samples of different syndiotacticity are shown in Fig. 13.21 Highly stereoregular and almost completely regioregular syndiotactic polypropylene samples, with concentration of the fully syndiotactic pentadrrrr in the range 75–96%, depending on the polymerization temperature and monomer concentration,2,21 and melting temperatures in the range 120–153 °C,21,26 are produced with ansa-zirconocenes with Cs symmetry (complexes 1–4 of Fig. 13).2 These catalysts produce highly syndiotactic polypropylene chains containing isolated mm triad stereodefects and isolated m diad defects.21
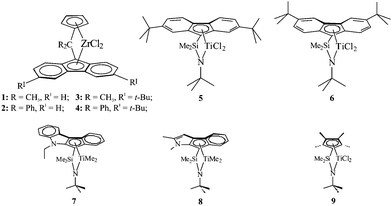 | ||
| Fig. 13 Structures of the Cs-symmetric zirconocene complexes (1–4) used as precursors of the catalysts for the polymerization of propene and the synthesis of highly syndiotactic polypropylene,2,21 and constrained geometry catalysts based on half-titanocene complexes (5–9) for the synthesis of poorly syndiotactic polypropylene.21,25,26,43,44 The catalyst 9 produces totally amorphous, moderately syndiotactic polypropylene.21,45 (Reproduced with permission from ref. 26. Copyright 2006 by the American Chemical Society). | ||
Lower syndiotacticity is achieved with the half-metallocene “constrained geometry” catalysts 5 and 6 of Fig. 13, with values of the rrrrpentad contents in the range 60–80% and melting temperatures in the range 80–120 °C.43 The constrained geometry catalysts containing a heterocycle condensed onto the cyclopentadienyl moiety (7 and 8 of Fig. 13) produce very poorly syndiotactic polypropylenes with concentration of the rrrrpentad of 40–55% and melting temperatures of 45–60 °C.44 Finally, the complexes precursors of the class of constrained geometry catalysts of formula Me2Si(Me4Cp)(NR)MtX2 (9 of Fig. 13), produce totally amorphous, moderately syndiotactic polypropylene with rrrr contents of about 20–25%.45
The values of melting temperature of the sPP samples prepared with the catalysts of Fig. 13 are shown in Fig. 14 as a function of the concentration of rrrrpentad.21,26 It is apparent that the stereoregularity and the melting temperature can be finely tuned in a wide range by changing catalyst structure and polymerization condition, while keeping high values of the molecular mass. In particular, very high values of molecular mass (even higher than one million) may be obtained for samples prepared with constrained geometry catalysts 5–8 of Fig. 13, which, indeed, show interesting mechanical properties even though the stereoregularity, the crystallinity and the melting temperatures are very low.21,25,26
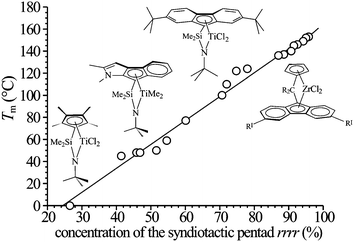 | ||
| Fig. 14 Melting temperatures of as-prepared samples of sPP having different stereoregularity, prepared with the different indicated catalysts, as a function of the concentration of the syndiotactic pentadrrrr.21,26 (Reproduced with permission from ref. 26. Copyright 2006 by the American Chemical Society). | ||
3.1 Influence of stereo-defects on the mechanical properties
The X-ray powder diffraction profiles and the stress-strain curves of compression-molded films of sPP samples having different stereoregularity and melting temperatures (Fig. 14), prepared with the different catalysts of Fig. 13, are shown in Fig. 15 and 16, respectively.21–26 The stress-strain curves of the nearly atactic, totally amorphous samples prepared with the catalyst 9 of Fig. 13 are also reported for comparison (Fig. 16C).21,25,26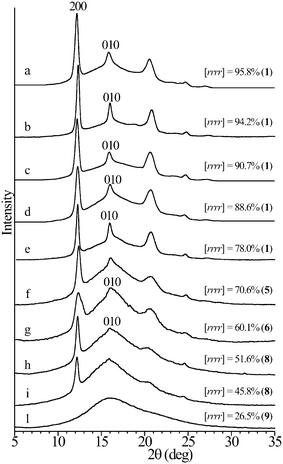 | ||
| Fig. 15 X-ray powder diffraction profiles of melt-crystallized compression molded films of some samples of sPP of different stereoregularity.21,22,25,26 The 200 and 010 reflections at 2θ = 12 and 16°, respectively, of the helical form I of sPP are indicated. The bold numbers in parenthesis indicate the catalysts of Fig. 13 used for the synthesis of the samples. Samples prepared with the same catalyst 1 show different stereoregularity since they have been synthesized at different polymerization temperatures.21 | ||
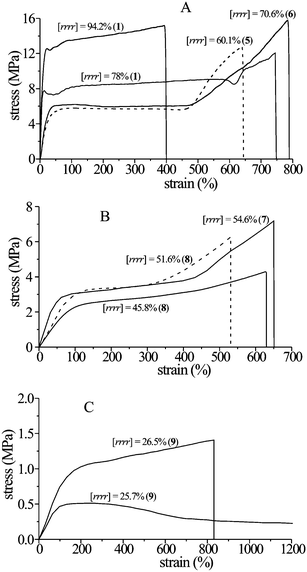 | ||
| Fig. 16 Stress–strain curves of unoriented compression molded films of some samples of sPP having different stereoregularity of Fig. 15.21,22,25,26 The bold numbers in parenthesis indicate the catalysts of Fig. 13 used for the synthesis of the samples. | ||
All samples crystallize from the melt in disordered modifications of the helical form I.21 The stress-strain data show that even highly stereoregular and crystalline sPP samples present high ductility and toughness at room temperature.26 The values of the Young's modulus, crystallinity and of the residual deformation (tension set) after breaking are reported in Fig. 17 as a function of stereoregularity (concentration of rrrrpentad). A decrease of the modulus and crystallinity and increase of ductility and flexibility with decreasing syndiotacticity is observed.21,22,25,26 The decrease of the values of tension set down to nearly 10% with decreasing stereoregularity and crystallinity (Fig. 17C) indicates that poorly stereoregular sPP samples show elastic behavior even in the form of unoriented compression-molded films.21,25,26 The origin of elasticity in sPP and the mechanism of plastic deformation have been extensively studied.21–28 The plastic deformation is associated with the occurrence of polymorphic transitions during stretching.21,23,26,27,28
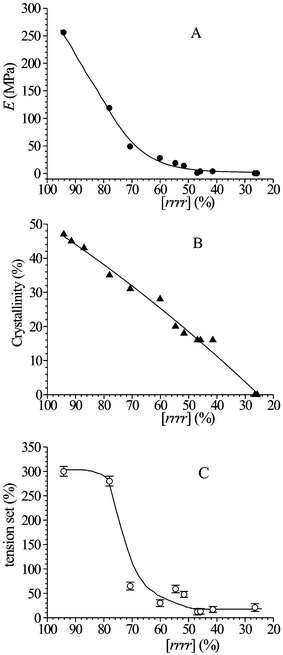 | ||
| Fig. 17 Values of the Young's modulus (A), X-ray crystallinity (B) and tension set after breaking (C) of unoriented compression-moulded films of samples of sPP of different stereoregularity, as a function of the concentration of rrrrpentad.21,26 | ||
Examples of X-ray diffraction patterns recorded during stretching of sPP samples are reported in Fig. 18 for samples of different stereoregularity, having [rrrr] = 91.5% (A–C) and 78% (A′–C′).27 For the more stereoregular samples, when a critical value of the strain εc = 90–100% is achieved, the stable form I with chains in two-fold helical conformation (Fig. 19A), present in the unoriented compression-molded films (Fig. 15), gradually transforms into the trans-planar form III (Fig. 18A–C). Fibers in the pure form III (Fig. 19B) are obtained only at higher deformation (Fig. 18C). The value of the critical strain εc increases with decreasing stereoregularity. In addition, different structural transformations are observed for samples of lower stereoregularity, having [rrrr] in the range 70–87% (Fig. 18A′–C′).21,27 The helical form I (Fig. 18A′) partially transforms into the trans-planar mesomorphic form upon stretching at deformations higher than the critical value εc (Fig. 18B′), and only at higher deformations the mesomorphic form transforms into form III (Fig. 18C′). For poorly syndiotactic samples prepared with constrained geometry catalysts 5–8 of Fig. 13, having rrrrpentad contents in the range 45–70%, the ordered trans-planar form III does not form by stretching even at very high deformation.25 The helical form I transforms at high deformations into the trans-planar mesomorphic form and the crystalline form III never forms.
![X-ray fiber diffraction patterns of fibers of sPP samples of different stereoregularity, with [rrrr] = 91.5% (A–C) and 78% (A′–C′), prepared with the catalyst 1 of Fig. 13, stretched at values of deformation ε. The 200 reflection of the helical form I and 020 and 110 reflections of the trans-planar form III are indicated.21,27](/image/article/2011/PY/c1py00129a/c1py00129a-f18.gif) | ||
| Fig. 18 X-ray fiber diffraction patterns of fibers of sPP samples of different stereoregularity, with [rrrr] = 91.5% (A–C) and 78% (A′–C′), prepared with the catalyst 1 of Fig. 13, stretched at values of deformation ε. The 200 reflection of the helical form I and 020 and 110 reflections of the trans-planar form III are indicated.21,27 | ||
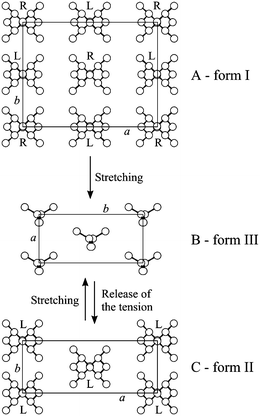 | ||
| Fig. 19 Crystal structures of the antichiral helical form I (A), the trans-planar form III (B) and the isochiral helical form II (C) of sPP. The trans-planar form III is obtained in oriented fibers by stretching unoriented samples in form I, and transforms into the helical form II by releasing the tension of the stretched fibers. | ||
The data in Fig. 18 have allowed building the phase diagram of sPP (Fig. 20), where the regions of stability of the different polymorphic forms of sPP in oriented fibers are defined as a function of stereoregularity and degree of deformation ε.
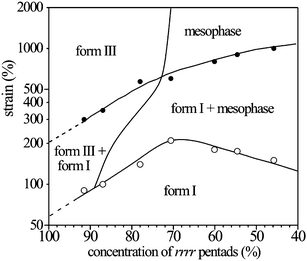 | ||
| Fig. 20 Phase diagram of sPP at room temperature as a function of strain and stereoregularity.27 | ||
The trans-planar form III obtained by stretching highly stereoregular samples is stable only in the stretched fibers and transforms into the helical form II upon releasing the tension or after breaking (Fig. 19B,C). This is shown by the X-ray fiber diffraction patterns of Fig. 21 of fibers of a highly stereoregular sPP sample stretched at high deformations (Fig. 21A), and after releasing the tension (Fig. 21B). The trans-planar form III obtained by stretching (Fig. 21A) transforms into the helical form II after relaxation by removing the tension (Fig. 21B).21–24,26,28
![X-ray fiber diffraction patterns of fibers of a highly stereoregular sample with [rrrr] = 94.2%, prepared with the catalyst 1 of Fig. 13, stretched at the indicated values of deformation ε, keeping the fiber under tension (A) and after the release of the tension (B).21,22,26 (Reproduced with permission from ref. 21. Copyright 2006 by Elsevier).](/image/article/2011/PY/c1py00129a/c1py00129a-f21.gif) | ||
| Fig. 21 X-ray fiber diffraction patterns of fibers of a highly stereoregular sample with [rrrr] = 94.2%, prepared with the catalyst 1 of Fig. 13, stretched at the indicated values of deformation ε, keeping the fiber under tension (A) and after the release of the tension (B).21,22,26 (Reproduced with permission from ref. 21. Copyright 2006 by Elsevier). | ||
During the transition from the two-fold helical form into the trans-planar form III by stretching (Fig. 19), an increase of the periodicity per structural unit h (which comprises two monomeric units) from h = c/2 = 3.7 Å of the two-fold helical form to h = c = 5.1 Å of the trans-planar form III, is involved. The crystal dimensions increase about 38% along c. This increase is completely recovered upon releasing the tension due to the transition of the trans-planar form into the helical form, and, correspondingly, a reduction of the length of the specimen occurs. As a result, for a not previously oriented material, a partial recovery of the macroscopic dimensions of the sample is attained.21–24,26,28
Oriented fibers of sPP show a perfect elastic recovery in successive cycles of stretching and relaxation, as shown by the hysteresis curves of Fig. 22, recorded for samples of different stereoregularity.21,22,24–26 As shown in Fig. 21, during these mechanical cycles a reversible crystal-crystal polymorphic transition between the helical form II and the trans-planar form III occurs.22–24,28 The helical form II of the stress-relaxed fibers transforms by stretching into the trans-planar form III, which transforms back into the helical form II by releasing the tension (Fig. 21 and 22). It has been demonstrated that this transition is a fast and direct process occurring on the same time scale as the rate the material is strained and relaxed.21,23,24,28 Since oriented fibers do not experience irreversible plastic deformation during the stretching steps of the mechanical cycles of Fig. 22, a complete elastic recovery is observed.
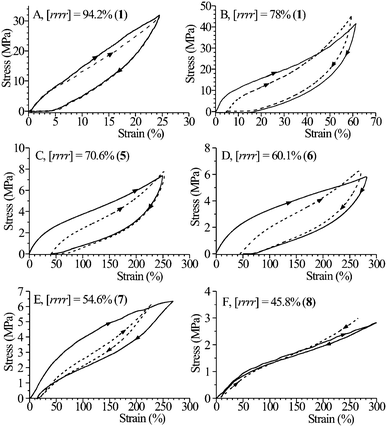 | ||
| Fig. 22 Stress-strain hysteresis cycles recorded at room temperature, composed of the stretching and relaxation (at controlled rate) steps according to the direction of the arrows, for strained and stress-relaxed fibers of some sPP samples having different stereoregularity. The stress-relaxed fibers have been prepared by stretching compression-molded films up to 400% (A, E, F) and 600% (B, C, D) deformations and then removing the tension. In the hysteresis cycles the stretching steps are performed stretching the fibers up to the final length Lf = 5L0 (A, E, F) and 7L0 (B, C, D), where L0 is the initial length of the unoriented compression-molded films. The first hysteresis cycle (continuous lines) and curves averaged for at least 4 cycles successive to the first one (dashed lines) are reported.21,22,25,26 The bold numbers in parenthesis indicate the catalysts of Fig. 13 used for the synthesis of the samples. | ||
It has been suggested that the polymorphic transition of the metastable trans-planar form III into the helical form II is in part responsible for the elasticity of sPP due to the enthalpy gain achieved when the fibers are relaxed.21–24,26,28 A model of “elasticity assisted” by the crystal-crystal phase transition has been suggested for the elastic behavior of sPP.22–24 In this idea, both the crystalline and amorphous chains play key roles. During elongation, the chains in the amorphous regions assume extended conformations and tend to orient parallel to the stretching direction. The crystalline aggregates also tend to assume a preferred orientation with the chain axes parallel to the stretching direction; at the same time, when a given crystal experiences a stress higher than a critical value a crystal–crystal phase transition from the helical form II into the trans-planar form III occurs and the size of the crystal increases by 38% along the chain axis direction. During the relaxation step, when a given crystal experiences a stress below a critical value, the trans-planar form III becomes unstable and transforms instantaneously into the more stable form II; correspondingly, the crystal shrinks by 38% along the chain axis direction.22–24 The enthalpy difference associated with the reversible crystal–crystal phase transition between the helical forms II and the trans-planar form III, ΔH(III→II) = −2.6 kJ mol−1 of monomeric units has been evaluated from the equilibrium values of the critical stress experienced by the crystals at various temperatures (found by stress-relaxation experiment) using a Clausius–Clapeyron type approach.28 The enthalpy gain achieved upon relaxation when the tensile tension is removed is mainly responsible for the elastic recovery of the sPP fiber samples.21–24,28
Upon relaxation the amorphous chains return to entropically favored disordered conformations, giving the entropic contribution to the elastic recovery process. Therefore, while the driving force leading the conventional elastomers to recover the initial dimensions is merely entropic, in the case of sPP elasticity is also assisted by the enthalpic gain achieved when the sample is relaxed.21–24,26 When the tension is removed, both the enthalpic factor, due to the structural transition in the crystalline regions, and an entropic factor, due to the conformational transition of the chains in the entangled amorphous phase, contemporarily contributes to the elastic recovery of the s-PP fibers.21–24,26
Remarkable differences in the elastic properties between highly stereoregular and crystalline sPP samples, prepared with Cs-symmetric catalysts 1–4 of Fig. 13, and the less syndiotactic samples and nearly amorphous samples, prepared with the constrained geometry catalysts 5–8 of Fig. 13, have been found.25,26 Poorly syndiotactic samples with rrrr concentration lower than 60–70%, show low Young's modulus and high ductility according to the low crystallinity (Fig. 16 and 17), and good elastic properties for both unoriented compression-molded films (Fig. 16B) and oriented fibers (Fig. 22C–F).25,26 During the mechanical cycles of Fig. 22C–F, the small and defective crystals of form I transform by stretching into the trans-planar mesomorphic form, which transforms back into the helical form I by releasing the tension.25,26 Therefore, for low syndiotactic and crystalline samples the elastic recovery is not associated with the reversible polymorphic transition between the trans-planar form III and the helical form II. This indicates that the enthalpic contribution to the elasticity plays progressively a less important role with decreasing stereoregularity and crystallinity. For these poorly syndiotactic samples the entropic effect of the conformational transition of the amorphous chains between disordered random-coil and extended conformations is mainly responsible for the elasticity.25,26
Fully amorphous samples nearly atactic samples prepared with the catalyst 9 of Fig. 13 present lower strength and experience rapid viscous flow of the chains at high deformations (Fig. 16C). In these samples only when the molecular weight is very high the viscous flow is prevented giving interesting elastic properties but, however, with very low strength.25
These data indicate that sPP shows different elastic behaviors depending on the stereoregularity, which can be controlled through the choice of the catalysts and conditions of polymerization. Thermoplastic elastomeric materials based on sPP with finely controlled physical and mechanical properties can be produced. As shown in Fig. 23, for the more syndiotactic samples having high melting temperatures (Fig. 14) and crystallinity, the elasticity has a mainly enthalpic character, due to the metastability of the trans-planar form III that transforms into the more stable helical form II. For less stereoregular samples, with low melting temperatures and crystallinity (Fig. 14 and 23), the elastic recovery is not associated with any polymorphic transitions and has a pure entropic origin, as in conventional elastomers. Depending on melting temperature and crystallinity, elastomers showing conventional entropic or unconventional enthalpic elasticity, can be obtained.21,26 The development of enthalpic elasticity allows maintenance of elastic properties even when the crystallinity, the melting temperature and the mechanical strength of the samples are very high. This is possible only thanks to the active role played by crystals in the elastic recovery.26
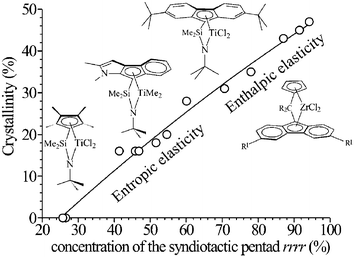 | ||
| Fig. 23 Enthalpic and entropic elasticity of sPP samples having different melting temperatures and crystallinity prepared with different catalysts. Highly syndiotactic samples with high melting temperatures and crystallinity show enthalpic elasticity, whereas samples with low stereoregularity, melting temperatures and crystallinity exhibit conventional entropic elasticity. | ||
Conclusions
The basic concept that the molecular structure of single macromolecules of polyolefins can be tailored through the design of the structure of metallorganic catalysts and can affect the crystallization behavior and the physical properties of the materials, has been clearly demonstrated in the case of isotactic and syndiotactic polypropylene. A fine tuning of the crystallization and physical properties of iPP and sPP-based materials is possible by tailoring the chain microstructure, through the controlled incorporation of stereo- and regio-defects, which can be achieved through the rational choice of the metallorganic complex precursor of the catalytic system. The random distribution of defects in metallocene-made polypropylene and the possible incorporation of a single type of defect has allowed understanding of the role played by microstructural defects on the properties of iPP and sPP.Novel iPPs having controlled desired properties, intermediate between those of stiff-plastic and elastomeric materials have been produced through the tailored design of new polymerization catalysts and the controlled incorporation of stereodefects. The mechanical behavior is strictly related to the presence of stereodefects and to the occurrence of polymorphic transformations during deformation. In the elastic iPPs, the elastic properties are associated with the occurrence of reversible phase transition that provides an enthalpic contribution to the elasticity.
Syndiotactic polypropylene shows elastomeric behavior, notwithstanding the high crystallinity and the relatively high glass transition temperature. Thermoplastic elastomeric materials based on sPP with finely controlled mechanical properties can be produced by tailoring the degree of stereoregularity. For more stereoregular samples with high melting temperatures and crystallinity, the elasticity has a mainly enthalpic character, due to the reversible phase transformation occurring during elastic recovery. This enthalpic contribution plays a progressively less important role with decreasing stereoregularity and crystallinity and for less syndiotactic samples the elastic recovery has a pure entropic origin. Depending on melting temperature and crystallinity, elastomers showing conventional entropic or unconventional enthalpic elasticity, can be obtained.
In conclusion, the case of polypropylene demonstrates the potential of metallorganic catalysts for the detailed study of properties of polymers in term of molecular architecture. Extension of this approach to other stereoregular polymers, such as isotactic polybutene, poly(4-methyl-1-pentene), syndiotactic polystyrene and corresponding copolymers, may contribute to clarify basic aspects of polymer physics related to the crystallization properties of polymers and the relationships between these properties and the material performance, and to identify the best molecular architecture and the synthetic pathway able to ensure the desired properties.
Acknowledgements
Financial support from Basell Polyolefins (Ferrara, Italy) is acknowledged. We thank Dr Luigi Resconi for the continuous collaboration and for having stimulated this work.Notes and references
- J. A. Ewen, J. Am. Chem. Soc., 1984, 106, 6355 CrossRef CAS.
- J. A. Ewen, R. J. Jones, A. Razavi and J. D. Ferrara, J. Am. Chem. Soc., 1988, 110, 6255 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS.
- W. Kaminsky, Macromol. Symp., 1998, 134, 63 Search PubMed.
- L. Resconi, R. L. Jones, A. Rheingold and G. P. A. Yap, Organometallics, 1996, 15, 998 CrossRef CAS.
- L. Resconi, F. Piemontesi, I. Camurati, O. Sudmeijer, I. E. Nifant'ev, P. V. Ivchenko and L. G. Kuz'mina, J. Am. Chem. Soc., 1998, 120, 2308 CrossRef CAS.
- L. Resconi, D. Balboni, G. Baruzzi, C. Fiori, S. Guidotti, P. Mercandelli and A. Sironi, Organometallics, 2000, 19, 420 Search PubMed.
- I. E. Nifant'ev, I. P. Laishevtsev, P. V. Ivchenko, I. A. Kashulin, S. Guidotti, F. Piemontesi, I. Camurati, L. Resconi, P. A. A. Klusener, J. J. H. Rijsemus, K. P. de Kloe and F. M. Korndorffer, Macromol. Chem. Phys., 2004, 205, 2275 CrossRef CAS.
- L. Resconi, S. Guidotti, I. Camurati, R. Frabetti, F. Focante, I. E. Nifant'ev and I. P. Laishevtsev, Macromol. Chem. Phys., 2005, 206, 1405 CrossRef CAS.
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587 CrossRef CAS.
- G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217 CrossRef CAS.
- G. W. Coates, Chem. Rev., 2000, 100, 1223 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- K. Angermund, G. Fink, V. R. Jensen and R. Kleinschmidt, Chem. Rev., 2000, 100, 1457 CrossRef CAS.
- G. Muller and B. Rieger, Prog. Polym. Sci., 2002, 27, 815 CrossRef CAS.
- C. De Rosa, F. Auriemma, A. Di Capua, L. Resconi, S. Guidotti, I. Camurati, I. E. Nifant'ev and I. P. Laishevtsev, J. Am. Chem. Soc., 2004, 126, 17040 CrossRef CAS.
- C. De Rosa, F. Auriemma, G. De Lucia and L. Resconi, Polymer, 2005, 46, 9461 Search PubMed.
- C. De Rosa, F. Auriemma, M. Paolillo, L. Resconi and I. Camurati, Macromolecules, 2005, 38, 9143 Search PubMed.
- C. De Rosa and F. Auriemma, J. Am. Chem. Soc., 2006, 128, 11024 CrossRef.
- C. De Rosa, F. Auriemma, O. Ruiz de Ballesteros, L. Resconi and I. Camurati, Chem. Mater., 2007, 19, 5122 Search PubMed.
- C. De Rosa and F. Auriemma, Prog. Polym. Sci., 2006, 31, 145 CrossRef CAS.
- F. Auriemma, O. Ruiz de Ballesteros and C. De Rosa, Macromolecules, 2001, 34, 4485 Search PubMed.
- F. Auriemma and C. De Rosa, J. Am. Chem. Soc., 2003, 125, 13143 Search PubMed.
- F. Auriemma and C. De Rosa, Macromolecules, 2003, 36, 9396 Search PubMed.
- C. De Rosa, F. Auriemma, O. Ruiz de Ballesteros, L. Resconi, A. Fait, E. Ciaccia and I. Camurati, J. Am. Chem. Soc., 2003, 125, 10913 CrossRef CAS.
- C. De Rosa, F. Auriemma and O. Ruiz de Ballesteros, Chem. Mater., 2006, 18, 3523 Search PubMed.
- C. De Rosa, F. Auriemma and O. Ruiz de Ballesteros, Phys. Rev. Lett., 2006, 96(16), 167801 Search PubMed.
- F. Auriemma, C. De Rosa, S. Esposito and G. R. Mitchell, Angew. Chem., Int. Ed., 2007, 46, 4325 Search PubMed.
- C. De Rosa, F. Auriemma and C. Perretta, Macromolecules, 2004, 37, 6843 CrossRef.
- F. Auriemma and C. De Rosa, Macromolecules, 2006, 39, 7635 Search PubMed.
- F. Auriemma, C. De Rosa and M. Corradi, Adv. Mater., 2007, 19, 871 Search PubMed.
- F. Auriemma, C. De Rosa, T. Boscato and P. Corradini, Macromolecules, 2001, 34, 4815 Search PubMed.
- C. De Rosa, F. Auriemma, T. Circelli and R. M. Waymouth, Macromolecules, 2002, 35, 3622 Search PubMed.
- F. Auriemma and C. De Rosa, Macromolecules, 2002, 35, 9057 Search PubMed.
- C. De Rosa, F. Auriemma and L. Resconi, Macromolecules, 2005, 38, 10080 Search PubMed.
- D. Fischer and R. Mülhaupt, Macromol. Chem. Phys., 1994, 195, 1433 CAS.
- R. Thomann, C. Wang, J. Kressler and R. Mülhaupt, Macromolecules, 1996, 29, 8425 CrossRef CAS.
- R. Thomann, H. Semke, R. D. Maier, Y. Thomann, J. Scherble, R. Mülhaupt and J. Kressler, Polymer, 2001, 42, 4597 Search PubMed.
- R. G. Alamo, M. H. Kim, M. J. Galante, J. R. Isasi and L. Mandelkern, Macromolecules, 1999, 32, 4050 Search PubMed.
- D. L. VanderHart, R. A. Alamo, M. R. Nyden, M. H. Kim and L. Mandelkern, Macromolecules, 2000, 33, 6078 Search PubMed.
- M. R. Nyden, D. L. Vanderhart and R. G. Alamo, Comput. Theor. Polym. Sci., 2001, 11, 175 Search PubMed.
- L. R. G. Treolar, The Physics of Rubber Elasticity, Oxford, Claderon Press, 1975 Search PubMed.
- A. Razavi and U. Thewalt, J. Organomet. Chem., 2001, 621, 267 Search PubMed.
- C. Grandini, I. Camurati, S. Guidotti, N. Mascellari, L. Resconi, I. E. Nifant'ev, I. A. Kashulin, P. V. Ivchenko, P. Mercandelli and A. Sironi, Organometallics, 2004, 23, 344 CrossRef CAS.
- J. C. Stevens, Stud. Surf. Sci. Catal., 1994, 89, 277 CAS.
| This journal is © The Royal Society of Chemistry 2011 |