Boron-containing polymers as versatile building blocks for functional nanostructured materials
Fei
Cheng
and
Frieder
Jäkle
*
Department of Chemistry, Rutgers University-Newark, 73 Warren Street, Newark, NJ 07102, USA. E-mail: fjaekle@rutgers.edu
First published on 18th May 2011
Abstract
This review aims to highlight recent advances in the emerging field of boron-containing polymers as precursors for nanostructured materials. Boron compounds display highly interesting physical and chemical properties, which are of great potential benefit in the field of materials science. Specifically, combination of the unique characteristics of boron-containing functional groups with the nano-size regime of dendritic structures and the self-assembly of polymers of well-defined architectures, such as telechelic polymers and block copolymers, allows scientists to develop next generation materials for sensing, electronic devices, catalysis, biomedical applications, etc.
 Fei Cheng | Fei Cheng graduated from Zhejiang University in 2005, earning a Bachelor in Polymer Materials and Engineering. He then moved to Fudan University for graduate studies, investigating the self-assembly of block copolymers and polymeric nanoparticles under the guidance of Prof. Daoyong Chen. After receiving a Master in Macromolecular Science in 2008, he joined the group of Prof. Jäkle at Rutgers-Newark for his PhD, working on the synthesis of luminescent boron containing polymers and nanostructured materials. |
 Frieder Jäkle | Frieder Jäkle is a Professor of Chemistry at the Newark Campus of Rutgers, the State University of New Jersey. He received his Diploma in 1994 and PhD in 1997 from TU München prior to joining Prof. Manners' group at the University of Toronto as a DFG postdoctoral fellow. In 2000 he started his independent research at Rutgers, where he was promoted to Associate Professor in 2006 and Full Professor in 2009. He is the recipient of several awards, including an NSF CAREER award (2004), an Alfred P. Sloan fellowship (2006), and a Friedrich Wilhelm Bessel Research Award of the Alexander von Humboldt Foundation (2009). |
Introduction
Bottom-up self-assembly of polymers, especially block copolymers, has been established as a powerful method for fabrication of nanostructured materials. With precise design and synthesis of polymeric building blocks and optimization of the self-assembly process, the size and morphology of self-assemblies can be controlled on the nanoscale.1 As a result, specific functional groups in the polymer building blocks can be placed at well-defined positions in the self-assembled nanospace. These are very important requirements for practical applications of functional polymers in solution, thin film and solid state.2An attractive new class of polymers receiving more and more interest recently contains boron functional groups as an integral part.3–5Boron is positioned just to the left of carbon in the periodic table and therefore contributes one less electron to its compounds. This leads to a number of interesting characteristics, including a tendency of tricoordinate boron species to act as Lewis acids toward a broad range of electron-rich substrates as well as to form conjugated π-systems that are often strongly colored and highly luminescent.6,7 Moreover, boron-containing compounds undergo a nuclear reaction upon exposure to neutrons, for which the 10B isotope has an exceptionally large cross-section. Thus, boron-containing polymers are promising for applications as supported catalysts, luminescent materials, biological imaging agents, chemical sensors and neutron detectors, stimuli-responsive materials, and as ceramic precursors.3–5
Taking advantage of the impressive achievements of controlled/living polymerization techniques,8 over the past several years the first boron-containing polymers with more complex architectures have emerged. In this review, we will highlight these new developments with a focus on the synthesis of boron-containing polymers of controlled architecture, their self-assembly in solution and bulk state, and their applications as new functional nanostructured materials. The discussion will cover different classes of polymers with functional groups ranging from boronic acids to ionic borates, borane and carborane clusters, and π-conjugated organoborane chromophores.
Boronic acid-containing polymers
It is well known that boronic acids can reversibly bind to sugars and other 1,2- or 1,3-diol compounds.9 As illustrated in Scheme 1 for phenylboronic acid (pKa ≈ 9)10 as a simple example, in basic solution the boronic acid group reacts with a diol, forming a boronic ester with a 5- or 6-membered ring. Neutral or acidic conditions typically result in hydrolysis of the boronic ester. On the other hand, in anhydrous organic solvents or in the presence of a Lewis base, trimerization leads to reversible formation of a boroxine ring with release of three water molecules.11 | ||
| Scheme 1 Boronic ester and boroxine formation. | ||
Polymers that contain boronic acid groups have a similar ability to bind to sugars and glycoproteins, and hence they have been extensively applied as supports in separation science and in the biomedical field.3 Successful introduction of boronic acid groups into block copolymer architectures on the other hand could open up new opportunities as a result of their self-assembly behavior. For instance, micellar or vesicular assemblies of boronic acid block copolymers are promising candidates for use in drug delivery vehicles and therapeutic agents. Boronic acid polymers are also expected to serve as versatile building blocks for stimuli-responsive self-assembled materials, given that boronic acids are weak electrolytes, whose solubility and charge state can be reversibly switched by pH changes. A challenge is, of course, to find suitable synthetic methods for the preparation of well-defined boronic acid-containing block copolymers. The reactive boronic acid groups tend to be not compatible with conventional living polymerization techniques, and in some cases, the direct polymerization of boronic acid monomers has been reported to result in cross-linked gels.12 To address these issues, boronic ester-containing polymers have been synthesized as precursors, whose deprotection gives the targeted boronic acid-containing polymers. More recently, the direct synthesis of boronic acid-containing polymers has also been realized by reversible addition–fragmentation chain transfer (RAFT) polymerization.
The first well-defined boron-containing block copolymer was reported by our group in 2005 (Scheme 2).13 Using atom transfer radical polymerization (ATRP), 4-pinacolatoborylstyrene was polymerized to give a product with narrow molecular weight distribution (PDI < 1.1). Chain extension with styrene led to a narrow block copolymer without any detectable amount of macroinitiator precursor or thermo-initiated polystyrene. ATRP of 4-pinacolatoborylstyrene with a poly(ethylene glycol) macroinitiator was more recently reported by the van Hest group.14 After deprotection of the pinacol ester group, the amphiphilic block copolymer poly(ethylene glycol)-b-poly(styrene boronic acid) (PEG-b-PSBA) was obtained. Vesicles were generated viaco-assembly of PEG-b-PSBA/PEG-b-PS in water (Fig. 1). When the PSBA weight fraction is lower than 10%, the PSBA forms evenly distributed domains in the continuous PS matrix. The PEG-b-PSBA can be removed from the vesicles by sugar binding at high pH, forming a permeable membrane. The vesicles were loaded with Candida AntarcticaLipase B (CALB), and used as nanoreactors for the hydrolysis of p-nitrophenyl acetate. The permeability and catalytic activity of these nanoreactors can be tuned through changes in the weight fractions of PSBA in the precursor.
 | ||
| Scheme 2 Synthesis of boronic ester block copolymer.13 | ||
 | ||
| Fig. 1 Proposed formation of vesicles with a permeable membrane. CALB (Candida AntarcticaLipase B) is incorporated into vesicles formed upon co-assembly of poly(ethylene glycol)-b-poly(styrene boronic acid) (PEG-b-PSBA) and PEG-b-PS in water. Addition of sugars leads to release of PEG-b-PSBA, which results in formation of permeable vesicles allowing substrates to enter and products to exit. Note that the PEG segments (blue) forming the outer layers of the vesicles are omitted in the schematic drawings. (Reproduced with permission from ref. 14. Copyright 2009 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
An alternative method for the preparation of boronic acid-functionalized block copolymers takes advantage of the selective exchange of the trimethylsilyl groups in species ArSiMe3 with BBr3.15 The amphiphilic block copolymer, poly(styreneboronic acid)-b-polystyrene (PSBA-b-PS), was obtained viasilicon–boron exchange and subsequent hydrolysis as illustrated in Scheme 3.13 The pH and solvent-dependent self-assembly of the block copolymer were studied by transmission electron microscopy (TEM) and dynamic light scattering (DLS).16 At high pH (0.1 M NaOH), the block copolymer forms spherical micelles (〈Dh〉 = 18 nm) of high interface curvature, due to strong electrostatic repulsion of the ionic R–B(OH)3−groups (Fig. 2, left). In contrast, at lower pH (0.001 M NaOH), the coexistence of neutral boronic acid groups RB(OH)2 reduces the curvature and leads to short worm-like structures (〈Dh〉 = 35 nm). In acetone/water and THF/water mixtures, other morphologies, including vesicles and larger compound micelles, could also be realized (Fig. 2, right).
 | ||
| Scheme 3 Synthesis of boronic acid block copolymers by post-modification.13 | ||
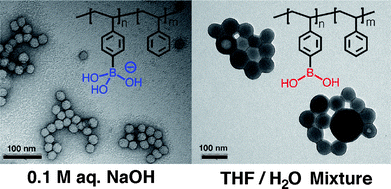 | ||
| Fig. 2 Self-assembled structures of PSBA-b-PS block copolymer in different solvents. (Reproduced with permission from ref. 16. Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
The Sumerlin group first introduced RAFT polymerization as a tool for the preparation of boron-containing polymers.17 A trithiocarbonate chain transfer agent (CTA) was used to control the polymerization of 4-pinacolatoborylstyrene. In the second step, the chain extension with N,N-dimethylacrylamide (DMA) afforded well-defined block copolymers. To generate the boronic acid block copolymer, the pinacolato ester group was quantitatively removed in a transesterification process that involved passing the polymer through a boronic acid-immobilized column.
Although boronic acid-containing polymers can be obtained by deprotection of boronic ester precursors, the direct polymerization of boronic acid monomers offers advantages such as lower cost and simpler operation. In more recent work, Sumerlin and co-workers directly polymerized the boronic acid monomer, 3-acrylamidophenylboronic acid (APBA), by RAFT polymerization and also prepared block copolymers with poly(N,N-dimethylacrylamide) (PDMA) and poly(N-isopropylacrylamide) (PNIPAM) as the second block.18,19 The PAPBA block establishes a pH-responsive equilibrium between neutral hydrophobic boronic acid and anionic hydrophilic boronate groups. The PAPBA polymer is also able to reversibly bind to diols, forming water-soluble ionic boronate ester species. Thus, in aqueous solution at high pH, the block copolymer is molecularly dissolved, whereas micelles with a PAPBA core and PDMA or PNIPAM shell form when the pH is below the pKa. Upon addition of glucose to the micelle solution at relatively lower pH, the micelles dissociate into single chains with formation of soluble boronate ester groups. For the PAPBA-b-PNIPAM block copolymer, temperature changes can be used as an additional stimulus. When the LCST of PNIPAM is reached, the block copolymer self-assembles into micelles with a PAPBA shell and PNIPAM core (Fig. 3). This multi-responsive behavior (pH, sugar, and thermal response) was monitored by DLS. Of particular interest is that the self-assembly of the block copolymers can be exploited for detection of sugars, where the sugar binding is visualized by disappearance of the light scattering that is characteristic of self-assembled micelles.
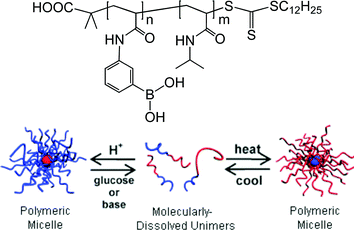 | ||
| Fig. 3 Self-assembly of PAPBA-b-PNIPAM in response to pH, sugar and temperature. (Adapted with permission from ref. 19. Copyright 2009 The Royal Society of Chemistry.) | ||
To enable applications of boronic acid polymers under physiological conditions, sugar binding has to occur at neutral pH. Toward this end, the van Hest group introduced a styryl monomer with a dialkylaminomethyl group adjacent to the boronic acid group (Wulff-type boronic acid).20 Lewis acid–base interaction between B and N lowers the pKa of the boronic acid, which favors sugar-binding at neutral pH. A block copolymer containing such a boronic acid-functionalized block in combination with a hydrophilic poly(ethylene glycol) (PEG) block was synthesized by RAFT polymerization using a PEG-based CTA (Fig. 4). In a buffer solution at neutral pH, the block copolymer micelles dissociate upon binding to monosaccharides.
 | ||
| Fig. 4 Sugar-binding to a Wulff-type boronic acid-containing block copolymer at neutral pH.20 | ||
Formation of borate and boronate ester links was also employed for surface coating, colloidal flocculation, and charging of the surface of colloidal particles.21–23 The Pelton group used phenylboronic acid-modified polyvinylamine (PVAm) to laminate cellulose films.21 The greatest adhesion was achieved at high pH with 150 kDa PVAm, with 16% of the amine groups bearing phenylboronic acid groups. In other work, they deposited polyol-stabilized PS latex particles on a boronic acid-derivatized regenerated cellulose surface.23 The deposition relies on the formation of boronate ester linkages. This process is reversible and the deposited particles can be released at pH 4. These reversible surface modifications based on borate and boronate ester formation provide an economical and convenient method to create surfaces that are biocompatible, feature designed patterns and contain other functionality.
As already noted above, in anhydrous organic solvents, boronic acids can reversibly form boroxines with elimination of three molecules of water (Scheme 1). Besides their more traditional applications, such as in flame retardants and as dopants for lithium ion batteries, recently, boroxine formation has been used to construct supramolecular tri-arm star polymers.11
The successful synthesis of telechelic boronic acid polymers is a key step in the preparation of boroxine-centered star polymers. Several methods have been developed. We used a silylated ATRP initiator to synthesize telechelic PS; silicon–boron exchange and subsequent hydrolysis gave the respective boronic acid-terminated polymer.24 Sumerlin and co-workers introduced RAFT polymerization with a boronic acid-functionalized CTA.25 The tri-arm star polymer with a boroxine core was then generated either by azeotropic distillation in an organic solvent24 or by addition of a base25 such as piperidine. When no base was added, the presence of water led to facile dissociation back into the boronic acid-terminated linear polymers. Similarly, in the case of the respective ditelechelic polymers, a cross-linked polymeric network can be generated reversibly.24
Very recently, the Iovine group demonstrated that boronic acid-terminated polycaprolactone (PCL) polymers can be prepared by ring-opening polymerization with a pinacol-protected initiator.26 The hydroxyl end group of PCL was then converted to an azide group, and further modified with various functional groups, such as phenyl, pyridyl, phenylboronic ester and zinc porphyrin. Deprotection of the pinacol ester led to a boronic acid group at the other end of the polymers. Star polymer formation was again achieved by addition of either pyridine or 7-azaindole as a Lewis base, D (Fig. 5). This approach allows for preparation of supramolecular star polymers with functional peripheral groups.
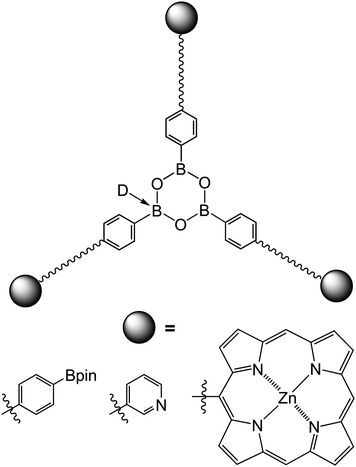 | ||
| Fig. 5 Functional supramolecular tri-arm star polymersviaboroxine formation.26 D = Lewis base. | ||
Conducting polymers that are functionalized with boronic acid groups are equally promising for applications in sensing and controlled release, and as building blocks for nanostructured materials.5,27 They have the potential benefit that electrochemical methods provide another tool for signaling in sensory applications. For instance, electrochemically polymerized poly(aniline boronic acid) (PABA) has been widely used as a potentiometric sensor for saccharides. Nanostructures based on PABA have been prepared by in situ chemical polymerization of aniline boronic acid in aqueous HCl or in aliphatic alcohols.28,29 Depending on the interaction between the boronic acid side groups and the solvents, Freund and co-workers obtained spherical nanoparticles, globular network, and nanofibers (Fig. 6). Polymer films produced from these nanostructures maintained high conductivity and showed enhanced redox stability. In another twist, the He group used an in situpolymerization method to prepare composites based on single-walled carbon nanotubes (SWNTs), single-stranded DNA (ssDNA), and PABA.30 The polymerization of 3-aminophenylboronic acid proceeded 4500 times faster in the presence of the ssDNA/SWNT composite, which was attributed to an interaction between the monomers and nanotubes. Moreover, the as-formed composites were shown to contain PABA of longer conjugation length. As a result, the composites are more stable and conductive.31 These boronic acid-containing conducting polymers and composites are of interest as sensors, coatings, and for flexible electronic devices.
 | ||
| Fig. 6 TEM images of poly(aniline boronic acid) (PABA) nanostructures prepared in different solvents and after precipitation and purification, redispersed in the same solvent: left: 0.1 M HCl, 2 h polymerization time; right: 1-propanol 5 h. (Adapted with permission from ref. 28. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Organoborate-containing polymers
While the reversible formation of anionic boronate species upon sugar binding or an increase in pH renders boronic acids hydrophilic and plays a key role in sensory applications, it is the inert nature of tetraarylborates that has led to widespread interest in these molecules as counterions to protect highly reactive organometallic catalysts, and as electrolytes in voltammetric applications and lithium ion batteries.32,33 To further enhance the stability and processability and to take advantage of confinement effects, the immobilization of organoborates onto polymeric scaffolds has been pursued by direct polymerization as well as post-modification methods.Cationic transition metal complexes play important roles in many catalytic processes and the nature of the counterion tends to critically affect the catalytic activity. For instance, metallocene complexes stabilized by weakly coordinating borate anions, e.g.tetrakis(pentafluorophenyl) borate ([B(C6F5)4]−), are widely used as highly active and selective catalysts for olefin polymerization.34 However, the reactive surface of traditional inorganic supports, such as silica or alumina, often is not very well compatible with these metallocene catalysts. To address this issue, Fréchet and co-workers developed a microsphere support in which ammonium functionalities were covalently attached through modification of a Merrifield-type PS resin; [B(C6F5)4]− served as the counterion.35Metallocene catalysts were then loaded onto the support by treating the borate-functionalized polystyrene beads with a toluene solution of the transition metal complex Cp2HfMe2. The highly swollen beads provide a catalytic environment close to that achieved in homogeneous catalysis, yet the low polarity of the interior of the beads ensures that the catalytic activity is retained. Copolymerization of ethylene and 1-hexene led to discrete spherical polyolefin beads with sizes of 0.3–1.4 mm. A conceptually different approach was taken by Uozumi, in which they covalently linked fluorinated borate moieties to PS beads for use in olefin polymerization.36
While the catalyst support represents a minor component and is therefore generally retained after formation of the polyolefin products, in other catalytic processes, the ability to easily recycle a heterogeneous catalyst is the primary motivation for immobilization. Using emulsion polymerization, Mecking and Vogt and co-workers prepared submicron PS particles that contain covalently linked tetraphenyl borate groups. They loaded the particles with a cationic rhodium catalyst by ion-exchange and used them as supported catalysts for hydrogenation reactions. Very low metal leaching was observed over several recycling processes.37
We introduced the first amphiphilic block copolymers with weakly coordinating borate groups covalently attached to the polymer, i.e. poly(styryltriphenylborate-b-polystyrene) (PSBPh3-b-PS) and poly(styryltris(pentafluorophenyl)borate)-b-polystyrene (PSBPf3-b-PS), via sequential ATRP and post-modification.38 The counterions of the borate block can be varied by ion exchange with sodium or tetrabutylammonium halides. In selective solvents such as water or methanol, the block copolymers form spherical micelles with a PS core and a poly(organoborate) corona. In contrast, in toluene, reverse micelles are obtained with an organoborate core and PS corona. The reverse micelles formed from PSBPf3-b-PS in toluene were loaded with Rh by treatment with the transition metal complex [Rh(cod)(dppb)]+(OTf)− (Fig. 7). The uptake of the Rh complex was confirmed by TEM. These weakly coordinating organoborate block copolymers are promising as nanoreactors for catalysis.
![Top: schematic illustration of organoborate block copolymer self-assembly in toluene (X = F, H). Bottom: TEM image of block copolymer micelles loaded with [Rh(cod)(dppb)]+. (Adapted with permission from ref. 38. Copyright 2010 American Chemical Society.)](/image/article/2011/PY/c1py00123j/c1py00123j-f7.gif) | ||
| Fig. 7 Top: schematic illustration of organoborate block copolymer self-assembly in toluene (X = F, H). Bottom: TEM image of block copolymer micelles loaded with [Rh(cod)(dppb)]+. (Adapted with permission from ref. 38. Copyright 2010 American Chemical Society.) | ||
Borate functionalities have been also attached to dendrimers. For instance, Mager et al. reported a series of weakly coordinating carbosilane dendrimers with organoborate functionalities at the periphery.39 The negatively charged dendrimers were used as cocatalysts for metallocene-catalyzed olefin polymerization. Ethene, propene, as well as ethene/propene and ethene/1-hexene mixtures were successfully polymerized. Importantly, the polymerizations could be performed in aliphatic solvents such as n-hexane, which is suitable for industrial production. Noteworthy is also that dendritic poly(pyrazolylborates) were used as scaffolds for other transition metals, such as rhodium or iridium.40
A different application of borate-containing polymers is as electrolytes for lithium ion batteries.41 Again, the weakly coordinating nature of borates is beneficial to minimize ion pairing that tends to reduce the lithium ion mobility. Typically, ethylene glycol or ethylene oxide segments are used to further facilitate transport of lithium ions through reversible binding to the ether functionalities. As an example, the Ohno group synthesized alkylborane and boric ester-type polymer electrolytes viahydroboration and dehydrocoupling polymerization of vinyl or hydroxyl-functionalized PEG oligomers and mesitylborane.42 The polymers show moderate ionic conductivities with relatively high selectivity for lithium ions. They also converted boric ester into borate-containing polymers by treatment with phenyl lithium to immobilize the ions onto the polymer chains. While the ionic conductivity of the borate type polymer was relatively low, a markedly higher lithium transference number was observed (tLi+ = 0.82–0.78). When pentafluorophenyl lithium or naphthyl lithium was used, a one order of magnitude increase in conductivity was achieved due to the improved dissociation of the lithium borate.
To apply borate counterions in nanostructured materials that display a bicontinuous network-like morphology is a promising approach to further increase the ion conductivity. Kato and co-workers recently introduced new types of ion conductive materials based on ionic liquids showing liquid crystalline bicontinuous cubic (LC Cubbi) phases (Fig. 8).43 The fan-shaped ionic liquid compounds feature an ammonium group at the focal point of the lipophilic alkylphenyl part, and a tetrafluoroborate counterion. These materials show excellent ion conductivity in the LC Cubbi state, which was attributed to the presence of 3-dimensionally interconnected channel networks. The Pulsed-Field-Gradient Nuclear Magnetic Resonance (PFG-NMR) technique was used to study the self-diffusion of ions in the confined nanospace. In the Cubbi state, a network of ion channels allows the anions to diffuse independently from the cations.44 Differently, in the isotropic phase, the anions and cations form pairs and/or clusters, and thus the materials show decreased ionic conductivity.
 | ||
| Fig. 8 Nano-ion channel network based on a LC bicontinuous cubic structure with Ia3d symmetry. (Adapted with permission from ref. 43. Copyright 2007 American Chemical Society.) | ||
The application of borate-containing polymers as active layers for electronic devices has been explored by the Bazan group.45 They fabricated a bilayer p–n junction by casting a cationic polyelectrolyte with fluoride counterions and a neutral conjugated polymer with dimesitylborane groups, which are known to be able to bind fluoride. An applied bias causes charge injection and fluoride ion migration from the polyelectrolyte layer to the neutral layer. Binding of fluoride to the boryl groups in the neutral layer leads to formation of the borate functionality. The original polyelectrolyte and neutral layers are therefore positively and negatively charged, respectively, leading to a p–n junction. The obtained device shows excellent light-emitting and current rectification performance (Fig. 9).
 | ||
| Fig. 9 Schematic illustration of a chemically fixed p–n junction (π-system = poly(fluorene-co-phenylene), PFP). (Adapted with permission from ref. 45. Copyright 2010 Macmillan Publishers Ltd. (Nature Materials).) | ||
The above examples demonstrate the applications of borate-containing polymers in catalysis, as ion conducting materials and in electronics. All these applications take advantage of the negative charge on the borate. It is reasonable to foresee growing interest also in the charge-reverse counterpart to the borate, the cationic boronium-containing polymers and nanomaterials. We recently reported the synthesis of the first organoboronium amphiphilic block copolymers with 2,2′-bipyridine as a strong donor ligand, and examined the self-assembly in methanol and toluene.46 The block copolymers form micellar structures in these selective solvents, and different counterions can be easily installed by ion exchange. The boronium-containing polymers are promising building blocks to construct composite materials with negatively charged dye molecules, polymers, nanoparticles, and biomolecules such as DNA.
Carborane-containing polymers
Borane and carborane clusters have over the years not only attracted broad interest because of their unusual 3-dimensional structure and non-classical bonding, but have also proven to be very useful building blocks for new materials. Several characteristic properties deserve special mention: first of all, the concentration of boron in these molecules is very high, making them superior candidates for applications in boron neutron capture therapy (BNCT) for cancer treatment;47 in addition, the high stability of carborane clusters makes them suitable for high temperature and chemically inert materials.48 Related is also the use of carborane anions as “chemical superweaklings”, in which the unusual cluster bonding (3D aromaticity) leads to effective delocalization of negative charge, thus enabling applications similar to those of weakly coordinating perfluoroarylborates discussed in the previous section.32,49Borane and carborane cluster-containing polymers have so far been most widely employed as precursors for boron carbide, boron nitride, and related ceramic materials.50Block copolymers that feature these clusters as building blocks are promising as precursors for porous ceramics, which in turn could serve as templates for nanomaterials synthesis, in gas storage and separation, and as supports for catalysts. Moreover, incorporation of carboranes into polymers and nanostructures is potentially beneficial for BNCT applications, where the size of the reagents greatly influences the ability to selectively deliver them to tumor cells. Applications as resists in microlithography51 and as materials with exceptional thermal stability are also envisioned.
In an effort at preparing nanostructured ceramic materials, Malenfant and co-workers synthesized an organic–inorganic block copolymer, polynorbornene-block-polynorbornenedecaborane (PNB-b-PDB), viaring-opening metathesis polymerization (ROMP) of a decaborane-functionalized norbornene monomer initially introduced by Sneddon and co-workers.52,53 Bulk self-assembly was achieved by dissolving the polymer in a good solvent followed by solvent evaporation.53 Interestingly, the polymer morphology depends on the solvent: when THF is used, a hexagonally packed cylindrical morphology with PNB cylinders and PDB as continuous phase is observed, while chloroform affords a lamellar structure. Upon pyrolysis in ammonia atmosphere, thin films cast from THF form a mesoporous boron nitride structure with highly ordered cylindrical morphology. The surface area is 950 m2 g−1, which is the highest to date for such a material. The pyrolysis of thin films cast from chloroform in nitrogen atmosphere leads to layered boron carbonitride/carbon ceramic composites (Fig. 10). In a complementary approach, Sneddon and co-workers prepared nanostructured ceramic materials by using a small molecule carborane precursor, 6,6′-(CH2)6–(B10H13)2, and alumina membranes or SiO2 colloidal crystals as template.54 Nanofibers, nanotubes and nanoporous materials were obtained after removal of the template.
 | ||
| Fig. 10 Self-assembly of a decaborane-functionalized block copolymer into cylindrical and lamellar morphologies and subsequent pyrolysis to form porous ceramic materials. (Adapted with permission from ref. 53. Copyright 2007 Macmillan Publishers Ltd. (Nature Nanotechnology).) | ||
The solution self-assembly of amphiphilic carborane-containing block copolymers was investigated by the Coughlin group.55 An amphiphilic block copolymer precursor was synthesized by ROMP, followed by deprotection of the second block. In water, the resulting block copolymer forms micelles with a carborane core, which are promising as delivery vehicles for BNCT. Carborane-containing block and random copolymers were also synthesized viaATRP of an acrylate-type carborane monomer and a poly(ethylene glycol) monomethyl ether methacrylate (MPEGMA).56 Both block and random copolymers show neutron capture property upon irradiation with thermal neutrons.
Instead of the synthesis of carborane-containing polymers, Matejicek and co-workers fabricated complex micelles by using a boron cluster [3-cobalt bis(1,2-dicarbollide)]− anion (CoD−) and a double hydrophilic block copolymer, poly(ethylene oxide)-block-poly(methacrylic acid) (PEO-b-PMA).57Hydrogen bonding and ionic interactions between CoD− and the PEO block lead to an insoluble NaCoD/PEO complex and ultimately to aggregation of the block copolymer. Nano-sized complex micelles with a PMA shell and a NaCoD/PEO core were observed by light scattering and electron microscopy.
Carborane-functionalized dendrimers (star-shaped macromolecules) represent another type of material with potential applications in medicine, catalysis, liquid crystalline substances, and thermally stable materials. Adronov and co-workers incorporated carborane units into aliphatic polyester dendrimers; fourth- and fifth-generation dendrimers with multiple carborane cages were prepared.58 Further modification of the periphery of the dendrimers with hydroxyl-terminated polyester afforded aqueous solubility. These carborane-containing dendrimers show boron neutron capture ability and thus serve as potential BNCT agents. Hosmane and co-workers developed a silicon tetrachloride mediated cyclotrimerization reaction of benzyl derivatives of carboranes to synthesize star-shaped molecules.59C3-Symmetric π-conjugated compounds were obtained, in which the peripheral carborane groups reduce π–π stacking interactions and thus enhance the fluorescence quantum yields. Deboronation of the o-carboranes led to water-soluble luminescent compounds. Higher generation dendrimers with up to 81 carborane cages in the periphery were prepared by Astruc and Hosmane and co-workers, who utilized azide–alkyne “click” coupling reactions to install the carborane moieties (Fig. 11). Formation of the carborane-functionalized dendrimers was confirmed by GPC and MALDI-TOF analysis and the hydrodynamic radius of the 81-cluster dendrimer was estimated by dynamic light scattering to be a remarkable Rh = 12.9 nm. The presence of the carborane moieties resulted in unusually high thermal stability.60
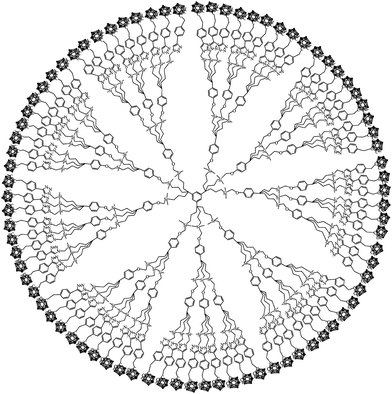 | ||
| Fig. 11 Schematic drawing of a dendrimer containing 81 carborane cages. (Adapted with permission from ref. 60. Copyright 2010 American Chemical Society.) | ||
Luminescent boron-containing polymers
Luminescent polymers play an important role in today's materials science, involving chemical sensors, optical and electronic devices, as well as imaging applications.61 Thousands of chromophores have been discovered and incorporated into polymeric structures over the past several decades. In very recent years, boron chromophores and luminescent boron-containing polymers have drawn particular interest, due to their facile synthesis, good stability, the broad selection of available ligands, tunable absorption and emission through the entire visible spectral window, as well as other novel photophysical properties, such as two-photon absorption, room-temperature phosphorescence and dual emission.5,7 From a structural point of view, luminescent polymers with boron chromophores in the main-chain, side-chain, or at the chain-end have been synthesized.The Fraser group used hydroxyl-functionalized difluoroboron dibenzoylmethane (BF2dbm) as initiator to polymerize lactide.62 Poly(lactic acid) (PLA) polymers with a luminescent BF2dbm end-group show interesting and unusual photophysical properties, namely, intense fluorescence, delayed fluorescence, two-photon absorption and oxygen-sensitive room-temperature phosphorescence (RTP). In dichloromethane solution, the polymer and initiator show similar absorption and emission. However, in the solid state, with an increase in molecular weight, the emission shifts from green to blue (507 to 443 nm). For the low molecular weight polymers, the concentration of BF2dbm is higher in the polymer matrix, and the excited state of BF2dbm is stabilized by chromophore–chromophore interactions. On the contrary, higher molecular weight polymers having lower BF2dbm content provide less stabilization of the excited state, and thus shorter wavelength emission is observed.63 Since the molecular weight can be well controlled by the polymerization time, this work presents a simple and effective color-tuning method, which can be used to generate molecular probes and sensors. In addition, because the thermal decay pathway from the triplet state is restricted by the polymer matrix, solid-state RTP was observed for BF2dbmPLA in the absence of oxygen. By using iodide-substituted difluoroboron dibenzoylmethane (BF2dbm(I)) as initiator, (BF2dbm(I))PLA with a high phosphorescence-to-fluorescence ratio was achieved.64
To investigate the biological applications of these polymers, Fraser and co-workers prepared boron-functionalized polylactide nanoparticles (BNPs) by adding the polymer solution into water.65 Biocompatible, water-soluble BNPs were obtained, which maintain the fluorescence, two-photon absorption and oxygen-sensitive room-temperature phosphorescence (RTP). The BNPs were successfully used to label chinese hamster ovary (CHO) cells (Fig. 12). Taking advantage of the dual-emissive and the oxygen-sensitive RTP properties, they also used the (BF2dbm(I))PLA and BNPs as oxygen sensor and imaging agent for tumor tissue.64 To enhance the stability of BNPs in biological conditions and facilitate the tumor uptake, they created complex nanoparticles with polyethylene glycol-block-poly(D-lactide) (mPEG-PDLA) and (BF2dbm(I))PLLA by co-precipitation, where the (BF2dbm(I))PLA and PDLA blocks form the core of the particles and the PEG block forms a water-soluble shell that stabilizes the complex BNPs.66 Besides, the synthesis and photophysical study of BF2dbm-terminated polycaprolactone (PCL) and PCL–PLA block copolymers were also reported.67
 | ||
| Fig. 12 Luminescence images of an aqueous suspension of BF2dbmPLA nanoparticles (left), and fluorescence and bright field microscopy image overlay of CHO cells incubated for one hour with a filtered BF2dbmPLA nanoparticle suspension (right). (Adapted with permission from ref. 65. Copyright 2008 American Chemical Society.) | ||
BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) chromophores possess high quantum yields, low rates of intersystem crossing, large molar absorption coefficients, and good photostability. They are used as biolabeling agents and for electronic devices.68 The Chujo group synthesized conjugated BODIPY-containing polymersviaSonogashira coupling reaction. A diiodo-substituted BODIPY and various aryl-diynes were used as building blocks (Fig. 13).69 Due to the π–π stacking interaction between BODIPY chromophores, the polymers form supramolecular aggregates in solution and the solid state. The π–π interactions can be modulated by using different aryl-diyne monomers, which was demonstrated by UV-vis characterization in chloroform. The π–π interactions also influence the aggregated structure of the polymer cast from THF. Weak interactions with 2,7-diethynyl-9,9-dihexyl-9H-fluorene give nano-sized particles, while stronger interactions with 1,4-diethynylbenzene as the building block result in nano-sized particles and higher-order fiber-like structures formed by aggregation of single particles.
 | ||
| Fig. 13 BODIPY polymer synthesis and SEM image of polymer with building block (b) dried at room temperature for 3.0 h on a glass plate. (Adapted with permission from ref. 69. Copyright 2008 American Chemical Society.) | ||
While the BODIPY chromophore is incorporated into the polymer main-chain in the previous example, Chujo and co-workers used RAFT polymerization to synthesize poly(methyl methacrylate) (PMMA) with pendant BODIPY groups.70 The BODIPY modified PMMA polymers were in turn used as macro-CTAs to control the polymerization of styrene. Well-defined homo- and block copolymers with BODIPY in the side chain were obtained. The authors suggest that π–π interactions between BODIPY groups in the side chain drive the self-assembly of homo- and block copolymers. The homopolymers form micrometre-sized blubber particles or chain-like structures. For the block copolymers, the PS shell prevents the formation of large aggregates, and nanoparticles with a PS shell were observed.
Using a BODIPY modified CTA, Chujo and co-workers also prepared telechelic PNIPAM polymers with a BODIPY end group.71In situreduction of HAuCl4 in the presence of PNIPAM results in gold nanoparticles (AuNPs) that are covered by PNIPAM, with the BODIPY groups located at the free end of the PNIPAM chains. The PNIPAM-coated AuNPs show thermo-responsive emission in water. Above the LCST, the PNIPAM polymers shrink towards the AuNP core. With the resulting decrease in the distance between BODIPY and AuNP core, a decreased emission from BODIPY was observed, which was attributed to fluorescence resonance energy transfer (FRET) and quenching due to BODIPY dye aggregation. The thermo-responsive emission of the BODIPY–PNIPAM modified AuNPs was reversible regardless of the number of heating/cooling cycles applied.
We reported the RAFT polymerization of luminescent boron quinolate monomers.72 Kinetic studies indicate pseudo first-order polymerization leading to narrow homopolymers. By using PEO-based macro-CTAs, amphiphilic block copolymers were obtained, which form micelles that exhibit good stability in aqueous solution (Fig. 14). The luminescence of polymer and micelle solutions can be tuned by using different quinolate ligands. The quinolate ligand affords green-emissive materials, while the respective ligand with an electron-donating arylamine substituent gives red-emissive materials, due to a charge transfer process.
 | ||
| Fig. 14 Amphiphilic organoboron quinolate block copolymers and images of their luminescence in aqueous solution. (Reproduced with permission from ref. 72. Copyright 2010 The Royal Society of Chemistry.) | ||
Conclusion and outlook
In light of their potential applications in catalysis, luminescent materials, chemical sensors, electronic devices, cancer therapy and other biomedical applications, nanomaterials derived from boron-containing polymers have become a new interest of chemists and material scientists. Future research on this topic is expected to involve:Development of new functional boron motifs
After more than a century, the chemistry of boron and its compounds is still a growing research area. Ever new boron-containing small molecules with interesting properties and functionalities are being discovered. As the selected examples in this brief review illustrate, incorporation of new boron moieties into polymers will lead to functional materials with a diverse range of desirable properties and new applications.Polymer synthesis
Direct polymerization of boron-containing monomers and post-modification procedures have been employed to synthesize boron-functionalized polymers. While in some cases, through highly efficient modification reactions, a general polymer precursor can produce a wide range of functional boron polymers, the advantages of direct polymerization methods lie in the broad applicability to numerous types of boron monomers and the ability to more easily realize well-defined polymer structures. Especially controlled free radical and living ring-opening polymerization techniques appear to be very promising routes to new boron-functionalized polymers.Self-assembly and materials fabrication
Polymer self-assembly in solution, thin film, and bulk state is widely used for fabrication of new materials. The examples discussed in this review demonstrate that through rational design of the polymer structure, self-assembled boron-containing materials with precisely controlled size, morphology, and functionality distribution can be realized. Besides, nanomaterial and surface chemistry are beginning to emerge as simple and efficient ways to prepare boron-functionalized nanoparticles and composite materials.As major achievements in terms of architectural control have been realized over the past just about five years and opened up a myriad of new applications of boron-containing polymers, this field is sure to attract more chemists, material scientists, physicists, and the industrial world.
Acknowledgements
We thank the National Science Foundation (CHE-0809642 and CHE-0956655) for financial support of our research programs on luminescent boron-containing polymers and the self-assembly of boron-containing block copolymers. F.J. is grateful to his current and former students and collaborators for their contributions to our research efforts in these areas; their names are cited in the pertinent references.References
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS; M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS; R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS; S. Park, D. H. Lee, J. Xu, B. Kim, S. W. Hong, U. Jeong, T. Xu and T. P. Russell, Science, 2009, 323, 1030–1033 CrossRef CAS; J. B. Gilroy, T. Gadt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS; C. J. Hawker and T. P. Russell, MRS Bull., 2005, 30, 952–966 CAS; R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS.
- F. Jäkle, J. Inorg. Organomet. Polym. Mater., 2005, 15, 293–307 CrossRef.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef; Y. Nagata and Y. Chujo, Macromolecules Containing Metal and Metal-Like Elements, in Boron-Containing Polymers, ed. A. S. Abd-El-Aziz, C. E. Carraher, Jr, C. U. Pittman, Jr and M. Zeldin, John Wiley & Sons, Inc., Hoboken, NJ, 2007, vol. 8, pp. 121–147 Search PubMed; N. Matsumi and Y. Chujo, Polym. J., 2008, 40, 77–89 Search PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; F. Jäkle, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley, Chichester, 2005, pp. 560–598 Search PubMed.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS; M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS; H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS; G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005 Search PubMed.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- K. Severin, Dalton Trans., 2009, 5254–5264 RSC; A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC.
- K. Kataoka, H. Miyazaki, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 1061–1062 CrossRef CAS; H. Ge, Y. Ding, C. Ma and G. Zhang, J. Phys. Chem. B, 2006, 110, 20635–20639 CrossRef CAS.
- Y. Qin, V. Sukul, D. Pagakos, C. Cui and F. Jäkle, Macromolecules, 2005, 38, 8987–8990 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- Y. Qin, G. Cheng, A. Sundararaman and F. Jäkle, J. Am. Chem. Soc., 2002, 124, 12672–12673 CrossRef CAS; Y. Qin, G. Cheng, O. Achara, K. Parab and F. Jäkle, Macromolecules, 2004, 37, 7123–7131 CrossRef CAS.
- C. Cui, E. M. Bonder, Y. Qin and F. Jäkle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2438–2445 CrossRef CAS.
- J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348–10349 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477–2479 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908–13909 CrossRef CAS.
- W. Chen, V. Leung, H. Kroener and R. Pelton, Langmuir, 2009, 25, 6863–6868 Search PubMed.
- R. Pelton, Z. Hu, H. Ketelson and D. Meadows, Langmuir, 2009, 25, 192–195 Search PubMed; R. Pelton, D. Zhang, K. L. Thompson and S. P. Armes, Langmuir, 2011, 27, 2118–2123 Search PubMed.
- D. Zhang, K. L. Thompson, R. Pelton and S. P. Armes, Langmuir, 2010, 26, 17237–17241 Search PubMed.
- Y. Qin, C. Cui and F. Jäkle, Macromolecules, 2007, 40, 1413–1420 CrossRef CAS.
- P. De, S. R. Gondi, D. Roy and B. S. Sumerlin, Macromolecules, 2009, 42, 5614–5621 CrossRef CAS.
- A. L. Korich, A. R. Walker, C. Hincke, C. Stevens and P. M. Iovine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5767–5774 Search PubMed.
- S. R. Ali, R. R. Parajuli, Y. Balogun, Y. Ma and H. He, Sensors, 2008, 8, 8423–8452 CrossRef CAS.
- B. A. Deore, I. Yu, J. Woodmass and M. S. Freund, Macromol. Chem. Phys., 2008, 209, 1094–1105 CrossRef CAS.
- B. A. Deore and M. S. Freund, Macromolecules, 2009, 42, 164–168 CrossRef CAS.
- Y. Ma, P. L. Chiu, A. Serrano, S. R. Ali, A. M. Chen and H. He, J. Am. Chem. Soc., 2008, 130, 7921–7928 CrossRef CAS.
- Y. Ma, W. Cheung, D. Wei, A. Bogozi, P. L. Chiu, L. Wang, F. Pontoriero, R. Mendelsohn and H. He, ACS Nano, 2008, 2, 1197–1204 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS.
- R. J. LeSuer and W. E. Geiger, Angew. Chem., Int. Ed., 2000, 39, 248–249 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS.
- S. B. Roscoe, J. M. J. Fréchet, J. F. Walzer and A. J. Dias, Science, 1998, 280, 270–273 CrossRef CAS.
- N. Kishi, C.-H. Ahn, J. Jin, T. Uozumi, T. Sano and K. Soga, Polymer, 2000, 41, 4005–4012 Search PubMed.
- R. Sablong, J. I. van der Vlugt, R. Thomann, S. Mecking and D. Vogt, Adv. Synth. Catal., 2005, 347, 633–636 CrossRef CAS.
- C. Cui, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2010, 132, 1810–1812 CrossRef CAS.
- M. Mager, S. Becke, H. Windisch and U. Denninger, Angew. Chem., Int. Ed., 2001, 40, 1898–1902 CrossRef CAS.
- J. A. Camerano, M. A. Casado, M. A. Ciriano and L. A. Oro, Dalton Trans., 2006, 5287–5293 RSC.
- N. Matsumi, K. Sugai, M. Miyake and H. Ohno, Macromolecules, 2006, 39, 6924–6927 CrossRef CAS; N. Matsumi, Y. Nakamura, K. Aoi, T. Watanabe, T. Mizumo and H. Ohno, Polym. J., 2009, 41, 437–441 CrossRef CAS; N. Matsumi, K. Sugai, K. Sakamoto, T. Mizumo and H. Ohno, Macromolecules, 2005, 38, 4951–4954 CrossRef CAS.
- N. Matsumi, K. Sugai and H. Ohno, Macromolecules, 2003, 36, 2321–2326 CrossRef CAS.
- T. Ichikawa, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2007, 129, 10662–10663 CrossRef CAS.
- A. E. Frise, T. Ichikawa, M. Yoshio, H. Ohno, S. V. Dvinskikh, T. Kato and I. Furó, Chem. Commun., 2010, 46, 728–730 RSC.
- C. V. Hoven, H. Wang, M. Elbing, L. Garner, D. Winkelhaus and G. C. Bazan, Nat. Mater., 2010, 9, 249–252 CAS.
- C. Cui, E. M. Bonder and F. Jäkle, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6612–6618 Search PubMed; C. Cui and F. Jäkle, Chem. Commun., 2009, 2744–2746 RSC.
- N. S. Hosmane, Y. Zhu, J. A. Maguire, S. N. Hosmane and A. Chakrabarti, Main Group Chem., 2010, 9, 153–166 Search PubMed; A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC.
- T. M. Keller, Carbon, 2002, 40, 225–229 Search PubMed.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS.
- Macromolecules Containing Metal and Metal-Like Elements, Boron-Containing Polymers, ed. A. S. Abd-El-Aziz, C. E. Carraher, Jr, C. U. Pittman, Jr and M. Zeldin, Wiley-Interscience, Hoboken, NJ, 2007, vol. 8 Search PubMed.
- J. Dai and C. K. Ober, Proc. SPIE, 2004, 5376, 508–516 Search PubMed , (Pt. 1, Advances in Resist Technology and Processing XXI).
- D. T. Welna, J. D. Bender, X. Wei, L. G. Sneddon and H. R. Allcock, Adv. Mater., 2005, 17, 859–862 CrossRef CAS.
- P. R. L. Malenfant, J. L. Wan, S. T. Taylor and M. Manoharan, Nat. Nanotechnol., 2007, 2, 43–46 CrossRef CAS.
- L. G. Sneddon, M. J. Pender, K. M. Forsthoefel, U. Kusari and X. Wei, J. Eur. Ceram. Soc., 2005, 25, 91–97 CrossRef CAS.
- Y. C. Simon, C. Ohm, M. J. Zimny and E. B. Coughlin, Macromolecules, 2007, 40, 5628–5630 CrossRef CAS.
- S. E. A. Gratton, M. C. Parrott and A. Adronov, J. Inorg. Organomet. Polym. Mater., 2005, 15, 469–475 Search PubMed.
- P. Matejıcek, J. Zednık, K. Uselova, J. Plestil, J. Fanfrlık, A. Nykanen, J. Ruokolainen, P. Hobza and K. Prochazka, Macromolecules, 2009, 42, 4829–4837 Search PubMed.
- M. C. Parrott, E. B. Marchington, J. F. Valliant and A. Adronov, J. Am. Chem. Soc., 2005, 2005, 12081–12089 Search PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS.
- R. Djeda, J. Ruiz, D. Astruc, R. Satapathy, B. P. Dash and N. S. Hosmane, Inorg. Chem., 2010, 49, 10702–10709 Search PubMed.
- S. W. Thomas, III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS; B. Z. Tang, Macromol. Chem. Phys., 2009, 210, 900–902 CrossRef CAS; A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS.
- G. Zhang, S. E. Kooi, J. N. Demas and C. L. Fraser, Adv. Mater., 2008, 20, 2099–2104 CrossRef CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS.
- A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS.
- F. R. Kersey, G. Q. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, ACS Nano, 2010, 4, 4989–4996 CrossRef CAS.
- G. Zhang, G. L. Fiore, T. L. St Clair and C. L. Fraser, Macromolecules, 2009, 42, 3162–3169 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- A. Nagai, J. Miyake, K. Kokado, Y. Nagata and Y. Chujo, J. Am. Chem. Soc., 2008, 130, 15276–15278 CrossRef CAS.
- A. Nagai, K. Kokado, J. Miyake and Y. Chujo, Macromolecules, 2009, 42, 5446–5452 CrossRef CAS.
- A. Nagai, R. Yoshii, T. Otsuka, K. Kokado and Y. Chujo, Langmuir, 2010, 26, 15644–15649 Search PubMed.
- F. Cheng and F. Jäkle, Chem. Commun., 2010, 46, 3717–3719 RSC.
| This journal is © The Royal Society of Chemistry 2011 |