Synthesis and photovoltaic properties of copolymers of carbazole and thiophene with conjugated side chain containing acceptor end groups
Zhi-Guo
Zhang
,
Haijun
Fan
,
Jie
Min
,
Siyuan
Zhang
,
Jing
Zhang
,
Maojie
Zhang
,
Xia
Guo
,
Xiaowei
Zhan
and
Yongfang
Li
*
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: liyf@iccas.ac.cn
First published on 28th April 2011
Abstract
Four alternating copolymers of carbazole (Cz) and thiophene (Th) with thienylene-vinylene (TV) conjugated side chains containing different acceptor end groups of aldehyde (PCzTh–TVCHO), mono-cyano (PCzTh–TVCN), di-cyano (PCzTh–TVDCN), and 1,3-diethyl-2-thiobarbituric acid (PCzTh–TVDT), have been designed and synthesized. The structures and properties of the main chain donor–side chain acceptor D–A copolymers were fully characterized. Through changing the acceptor group attached to the TV conjugated side chain on thiophene unit, the electronic properties and energy levels of the copolymers were effectively tuned. The effect of substituent on the electronic structures of the copolymers was also studied by molecular simulation. These results indicate that it is a simple and effective approach to tune the bandgap in a conjugated polymer by attaching an acceptor end group on the conjugated side chains. The polymers were used as donor in polymer solar cells. The device based on PCzTh–TVDCN/PC70BM demonstrates a power conversion efficiency of 2.16% with a high Voc of 1.03 V under the illumination of AM1.5G, 100 mW cm−2.
1. Introduction
Polymer solar cells (PSCs) are considered as a promising alternatives to inorganic semiconductor-based solar cells for the generation of affordable, clean, and renewable energy.1 Advantages of the PSCs include low-cost fabrication of large-area devices, light weight, mechanical flexibility, and easy tunability of chemical properties of the polymer materials. Therefore, there has been great interest from academia and application-oriented companies to increase the efficiency of PSCs for future commercial application, primarily through new polymer donors2–5 and fullerene derivative acceptors6 development and device engineering.7,8 PSCs are commonly composed of a blend film of conjugated polymer donor and soluble fullerene derivative (such as PCBM) acceptor sandwiched between an ITO positive electrode and a low workfunction metal negative electrode. It has been realized that an ideal polymer donor in PSCs should exhibit broad absorption with high absorption coefficient in the visible region, high hole mobility, suitable energy level matching to the fullerene acceptor, and appropriate compatibility with fullerene acceptor to form nanoscale bicontinuous interpenetrating network.9 All these specific design criteria can offer high values of short-circuit current (Jsc), open-circuit voltage (Voc) and fill-factor (FF) of the PSCs, all of which are related to the power conversion efficiency (PCE) by the following equation: PCE = (Jsc × Voc × FF)/Pin, where Pin is the input light power.One feasible approach towards broadening the visible absorption and tuning the energy levels is to design alternating donor–acceptor (D–A) copolymers, in which the hybridization of HOMO located on the donor moiety with LUMO located on the acceptor moiety provides a means for narrowing the bandgap and tuning the HOMO and LUMO energy levels of conjugated polymers.10–12 Based on this approach, main chain D–A copolymers have witnessed great success in achieving high performance PSCs.13–25
Previous studies in our group have developed a new family of two-dimensional conjugated polymers with conjugated side chains.5,26 This type of polymers features high hole mobility thanks to the overlapping of the side chain interactions with the conjugated main chains, and broad absorptions deriving from both the main chains and side chains, thus demonstrated prominent device performances in PSCs26a and OFETs (organic field-effect transistors)27,28 Recently, Huang et al. extended the concept of two-dimensional conjugated polymers to the copolymers of fluorene and triphenylamine (TPA) with thienylene-vinylene conjugated side chain attaching an electron-withdrawing end group.29 Subsequently, the new D–A copolymers with main chain donor containing triarylamine unit with acceptor end groups at conjugated side chains attracted much attention for application in PSCs.29–32 Furthermore, we synthesized a new main chain donor-side chain acceptor D–A copolymer by the copolymerization of fluorene and thiophene unit instead of TPA in the main chain recently, for improving the planarity of the polymer main chain.33
Poly(2,7-carbazole) derivatives are among the most promising conjugated polymers used in fabricating the optoelectronic devices.34,35 They possess the advantages of high hole mobility, good solution processability, deeper HOMO energy level and air stable features, deriving from the natures of the carbazole unit, such as rigid biphenyl unit, delocalization of the lone electron pair of nitrogen atom over aromatic structure and the bulky alkyl chains attached. The alternating copolymers of 2,7-carbazole and various acceptors have been synthesized for applications as the donor materials in PSCs.36,37 The combination of the unique features of the 2,7-carbazole and the advantages of the main chain donor-side chain acceptor architecture may offers some promising donor materials for high-performance PSCs.
Based on the consideration mentioned above, alternating copolymers of 2,7-carbazole (Cz) and thiophene (Th) with thienylene-vinylene (TV) conjugated side chain containing various acceptor end groups, such as aldehyde (PCzTh–TVCHO), mono-cyano (PCzTh–TVCN), di-cyano (PCzTh–TVDCN), and 1,3-diethyl-2-thiobarbituric acid (PCzTh–TVDT), were designed and synthesized in this work, for application as donor materials in PSCs. Also, the effect of pendant acceptors on absorption, morphology, mobility, and photovoltaic properties of the copolymers was fully investigated.
2. Experimental section
2.1 Instrumentation
1H NMR and 13C NMR spectra were measured on a Bruker DMX-400 spectrometer with d-chloroform as the solvent and tetramethylsilane as the internal reference. UV–visible absorption spectra were measured on a Hitachi U-3010 UV–vis spectrophotometer. Photoluminescence spectra were measured using a Hitachi F-4500 spectrophotometer. Fourier transform infrared (FT-IR) spectroscopy was performed on a Shimadzu FTIR-8400 spectrophotometer. A mixture containing 100 mg of potassium bromide (KBr) and 1–2 mg of the polymer was ground into a fine powder and compressed to form a transparent pellet for the FT-IR measurement. Melting points were determined on a WRS-1B Digital Melting Point Apparatus and were uncorrected. Mass spectra were recorded on a Shimadzu spectrometer. Elemental analyses were carried out on a flash EA 1112 elemental analyzer. Thermogravimetric analysis (TGA) was conducted on a Perkin-Elmer TGA-7 thermogravimetric analyzer at a heating rate of 20 °C min−1 and under a nitrogen flow rate of 100 mL min−1. Gel permeation chromatography (GPC) measurements were conducted, using polystyrene as standard and THF as the eluent. The electrochemical cyclic voltammetry was performed on a Zahner IM6e Electrochemical Workstation, in a 0.1 mol L−1acetonitrile solution of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) at a potential scan rate of 100 mV s−1 with an Ag/Ag+ reference electrode and a platinum wire counter electrode. Polymer film was formed by drop-casting 1.0 μL of polymer solutions in THF (analytical reagent, 1 mg mL−1) onto the working electrode, and then dried in the air.2.2 Photovoltaic device fabrication and characterization
The PSCs were fabricated with a configuration of ITO/PEDOT:PSS (40 nm)/active layer (40–90 nm)/Al (40 nm) or Ca(8 nm)/Al(40 nm). A thin layer of PEDOT:PSS (poly-(3,4-ethylenedioxythiophene): poly(styrene sulfonate)) was spin-cast on pre-cleaned ITO-coated glass from a PEDOT:PSS aqueous solution (Baytron P VP AI 4083 from H. C. Starck) at 2000 rpm and dried subsequently at 150 °C for 30 min in air, then the device was transferred to a glovebox, where the active layer of the blend of the copolymers and fullerene acceptors was spin-coated onto the PEDOT:PSS layer. Finally, a metal top electrode was deposited in vacuum onto the active layer at a pressure of ca. 5 × 10−5 Pa. The active area of the device was ca. 4 mm2.The thickness of the active layer was determined by an Ambios Tech. XP-2 profilometer. The current–voltage (I–V) characteristics were measured on a computer-controlled Keithley 236 Source-Measure Unit. A xenon lamp coupled with AM 1.5 solar spectrum filter was used as the light source, and the optical power at the sample was around 100 mW cm−12.
The morphology of blend film was observed by an atomic force microscopy (AFM) (NanoMan VS, Veeco, USA) in contact mode. Charge carrier mobility in blend films was measured by space charge limited current (SCLC) method. The hole-only devices with the same active layer thickness as actual devices were fabricated with an ITO/PEDOT:PSS/photoactive layer/Au structure. The hole mobility was calculated by fitting the dark J–V curves for the hole-only devices to SCLC model at low voltages, in which the current density is given by J = 9ε0εrμV2/8L3exp[0.891γ(V/L)0.5], where ε0εr represents the permittivity of the material, μ is the mobility, γ is the field activation factor, and L the thickness of the active layer. The applied bias voltage is corrected for the built-in potential so that V = Vapplied − Vbi.
2.3 Materials
Diethyl (2,5-dibromothiophen-3-yl) methylphosphonate26 (1) and 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)-N-9′-heptadecanylcarbazole31 (4) were prepared according to the method reported previously. Tetrakis(triphenylphosphine)palladium, Trioctylmethylammonium chloride (Aliquat® 336), octyl cyanoacetate, malononitrile and 1,3-diethyl-2-thiobarbituric acid were purchased from Sigma-Aldrich Chemical Co. The solvents (pyridine, DMF and 1,2-dichloroethane) were dried by standard procedure and distilled before use. The synthesis routes for the copolymers are shown in Schemes 1 and 2.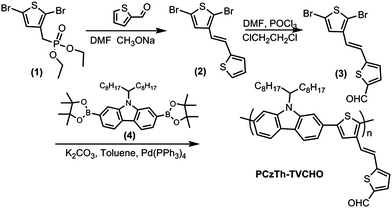 | ||
| Scheme 1 Synthesis route of PCzTh–TVCHO. | ||
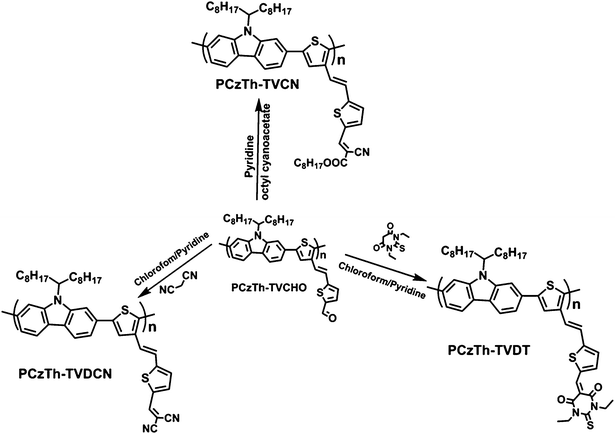 | ||
| Scheme 2 Synthesis route of PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) yielded 3 as yellow crystals (3.2 g, 94%). GC-MS: m/z = 378. 1H NMR (400 MHz, CDCl3, δ, ppm): 9.87 (S, 1H), 7.67 (m, 1H), 7.17(m, 2H) 6.98(m, 2H).
:1) yielded 3 as yellow crystals (3.2 g, 94%). GC-MS: m/z = 378. 1H NMR (400 MHz, CDCl3, δ, ppm): 9.87 (S, 1H), 7.67 (m, 1H), 7.17(m, 2H) 6.98(m, 2H).
3. Results and discussion
Polymer synthesis and characterization
Chemical structures and synthetic routes of the main chain donor-side chain acceptor D–A copolymers are depicted in Schemes 1 and 2. Initially, Horner–Wordsworth–Emmons (HWE) reaction was performed between phosphonate precursor (1) and thiophene-2-carbaldehyde (2), affording intermediate (3). The conformation of the vinylic bonds of compound (2) have unequivocally been designated as E on the basis of the JHH vinylic coupling constant (ca. 16 Hz). Subsequent treatment of compound (2) with DMF and POCl3via the Vilsmeier reaction afford dibromide (3) featuring thienylene-vinylene bridged aldehyde group.The dibronate 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2-yl)-N-9′-heptadec anylcarbazole (4) was synthesized from commercially available 2,7-carbazolevia straightforward procedure recently.31
The “basic” copolymer, viz. PCzTh–TVCHO, was synthesized by Suzuki polycondensation of an equimolecular mixture of the dibromide (3) and diboronate (4), as illustrated in Scheme 1. The copolymerization was carried out for 48 h using Pd(PPh3)4 as the catalyst in a two-phase mixture of degassed toluene and aqueous K2CO3 (2.0 M) and in the presence of Aliquat 336 as the phase transfer reagent. To study the structure–properties relationships, polymers with similar molecular architecture and bearing comparable numbers of conjugation units are more desirable.38 To overcome the reactivity difference among the monomers, a post-modification approach was adopted to modify the pendant groups on the thienylene–vinylene side chains of PCzTh–TVCHO, forming PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT. Thus, all the three polymers have the same number of conjugation units as PCzTh–TVCHO with fine-tuned electronic structure.
As shown in Scheme 2, for PCzTh–TVCN, the optimum reaction conditions to achieve complete conversion of the aldehyde groups was found to be at 55 °C for 48 h in pyridine as both solvent and base, while for PCzTh–TVDCN and PCzTh–TVDT, the optimum reaction conditions are in chloroform at 40 °C for 24 h and using 1 mL pyridine as base. After purification and drying, PCzTh–TVCHO, PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT were obtained as yellow, yellowish-red, red and purple solids, respectively, indicating a significant change in their optical properties. The bulky alkyl side chains attaching on the nitrogen atom of the carbazole unit combined with the two-dimensional architecture, account for the high solubility of the copolymers in chlorinated solvents, such as dichlomathane, chloroform and dichlorobenzene, which is important for their application in PSCs.
The conversion of aromatic aldehyde of PCzTh–TVCHO to other acceptor groups was confirmed by Fourier transform infrared (FT-IR) absorption spectroscopy. As shown in Fig. 1, the characteristic absorption peaks of the –CHO group appeared at 1666 and 2796 cm−1, belonging to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H stretching vibrations, respectively.10After transformation into PCzTh–TVCN and PCzTh–TVDCN, these characteristic absorptions of aldehyde disappeared, and new absorption peaks at 2217 cm−1 (corresponding to C
O and C–H stretching vibrations, respectively.10After transformation into PCzTh–TVCN and PCzTh–TVDCN, these characteristic absorptions of aldehyde disappeared, and new absorption peaks at 2217 cm−1 (corresponding to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching for the two polymers) and 1710 cm−1 (corresponding to CO stretching for PCzTh–TVCN) emerged, indicative of complete conversion of the terminal aldehyde group. It should be noted that for PCzTh–TVDT, due to overlapping of vibration in the aldehyde group and that of 1,3-diethyl-2-thiobarbituric acid moiety, the conversion of aldehyde group into the 1,3-diethyl-2-thiobarbituric acid group in PCzTh–TVDT cannot be identified in the FT-IR spectrum. However, it can be clearly confirmed in the 1H NMR spectra. In the 1H NMR spectra, the complete disappearance of the aldehyde proton signal at 9.8 ppm31 and the appearance of olefinic proton signals at 8.26 ppm for PCzTh–TVCN, 7.74 ppm for PCzTh–TVDCN and 8.62 ppm31 for PCzTh–TVDT, provide confirmation of the complete structural conversion. In addition, the ratios of integrated peak areas of olefinic to aliphatic protons and that of olefinic to methine group adjacent to the carbazoleN atom protons are consistent with the proposed structures of PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT, as depicted in Scheme 2.
N stretching for the two polymers) and 1710 cm−1 (corresponding to CO stretching for PCzTh–TVCN) emerged, indicative of complete conversion of the terminal aldehyde group. It should be noted that for PCzTh–TVDT, due to overlapping of vibration in the aldehyde group and that of 1,3-diethyl-2-thiobarbituric acid moiety, the conversion of aldehyde group into the 1,3-diethyl-2-thiobarbituric acid group in PCzTh–TVDT cannot be identified in the FT-IR spectrum. However, it can be clearly confirmed in the 1H NMR spectra. In the 1H NMR spectra, the complete disappearance of the aldehyde proton signal at 9.8 ppm31 and the appearance of olefinic proton signals at 8.26 ppm for PCzTh–TVCN, 7.74 ppm for PCzTh–TVDCN and 8.62 ppm31 for PCzTh–TVDT, provide confirmation of the complete structural conversion. In addition, the ratios of integrated peak areas of olefinic to aliphatic protons and that of olefinic to methine group adjacent to the carbazoleN atom protons are consistent with the proposed structures of PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT, as depicted in Scheme 2.
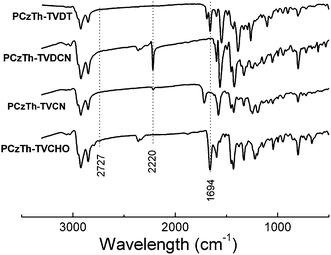 | ||
| Fig. 1 FT-IR spectra of the copolymers. | ||
Molecular weight of the polymers was determined by gel permeation chromatography (GPC). Number-average molecular weight (Mn) of PCzTh–TVCHO is 1.26 × 104, corresponding to a degree of polymerization of about 20, with a polydispersity index of 3.84. The structural transformation of PCzTh–TVCHO into the other three copolymers has led to a slight increase in molecular weights of the resulting copolymers, as shown in Table 1. The Mns of PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDCN were found to be 1.60 × 104, 1.38 × 104, and 1.59 × 104 g mol−1 respectively, with the corresponding polydisperisty indices of 3.72, 3.83, and 3.59.
| polymers | M n (g/mol) | PDI (Mw/Mn) | T d a(°C) | λ abs b (nm) | E g c (eV) | HOMOd (eV) | LUMOe (eV) |
|---|---|---|---|---|---|---|---|
| a 5% weight loss temperature measured by TGA under N2. b For the polymer films. c Band gap, estimated from the optical absorption band edge of the films. d Calculated from the onset oxidation potentials of the polymers. e Estimated using empirical equations ELUMO = EHOMO + Eg. | |||||||
| PCzTh–TVCHO | 12.6 K | 3.84 | 378 | 394 | 2.46 | −5.56 | −3.10 |
| PCzTh–TVCN | 16.0 K | 3.72 | 185 | 408, 509 | 2.18 | −5.57 | −3.39 |
| PCzTh–TVDCN | 13.8 K | 3.83 | 292 | 412, 521 | 2.05 | −5.60 | −3.55 |
| PCzTh–TVDT | 15.9 K | 3.59 | 398 | 418, 537 | 1.94 | −5.59 | −3.65 |
Thermal stability
The thermal properties of the copolymers were determined viathermogravimetric analysis (TGA) under a nitrogen atmosphere. As shown in Table 1 and Fig. 2, the 5% weight-loss temperatures for PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT are in the range of 185–398 °C, which is adequate for application of the copolymers as active materials in the optoelectronic devices.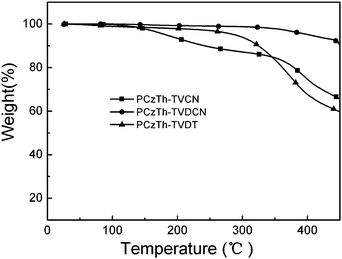 | ||
| Fig. 2 TGA plots of the copolymers with a heating rate of 10 °C min−1 in an inert atmosphere. | ||
Optical properties
Fig. 3 shows the absorption spectra of the copolymers in solution and in the solid films. In the polymer solutions, the maximum absorptions of the copolymers at 400 nm for PCzTh–TVCHO, 412 nm for PCzTh–TVCN, 413 nm for PCzTh–TVDCN and 415 nm for PCzTh–TVDT (Fig. 3(a)), correspond to the π–π* transition of the polymer backbone. The π–π* transition absorption peaks of the polymer backbones, are slightly red shifted when the pendant acceptor groups vary from the aldehyde group to the mono-cyano group, to the dicyano group and further to 1,3-diethyl-2-thiobarbituric acid group. This phenomenon indicates that the π-conjugated backbone of the polymers is conjugated with the pedant π-bridge and the red-shifted extension of absorption is associated with the extension of the effective conjugation length between the polymer main chain and pedant π-bridge10 The molar extinction coefficients per repeating unit of PCzTh–TVDCN at peak wavelength is 5.07 × 104 M−1 cm−1, higher than that of PCzTh–TVCN (4.70 × 104 M−1 cm−1) and PCzTh–TVDT (4.90 × 104 M−1 cm−1).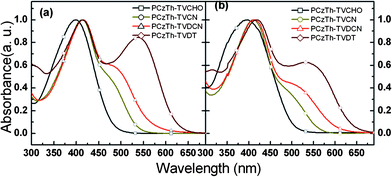 | ||
| Fig. 3 UV-visible absorption spectra of the copolymers (a) in toluene solution and (b) in film. | ||
Differing from PCzTh–TVCHO and in addition to the absorptions of Cz-alt-Th backbones, the introduction of electron-withdrawing pendant groups produces new absorption bands at 479 nm for PCzTh–TVCN, 491 nm for PCzTh–TVDCN and 540 nm for PCzTh–TVDT, attributable to the intramolecular charge transfer (ICT) interactions10,38 between the polymer backbone and the pendant acceptor groups. Thus, both the main chain and side chain contribute to the broad nature of the absorptions for the copolymers, which is much like that of the conjugated side chain polymers developed in our group.26 Evidently, the ICT interactions are progressively enhanced with the increase of the electron affinity of the pendant acceptor group in the D–A copolymers, due to the enhanced electron accepting ability of the acceptor moiety and enlarged conjugation path.10 For the as-spun film on quartz glass substrates, the UV–visible absorption spectra of the copolymers are red-shifted and broadened (Fig. 3(b)), as compared with that of their corresponding dilute solutions. The red-shift and broadening of the absorption spectra of the polymer films is a common phenomenon, which results from the enhanced interchain interaction in the solid films and is probably related to increased polarizability of the film.10 It seems that the interchain interaction in the main chain donor-side chain acceptor D–A copolymers is not as strong as those of the main chain D–A copolymers, probably due to the fact that the bulky side chains of the Th moiety retard the conformation change of the polymer main chains in the solid state.39 The absorption edges of the copolymer films are located at 505 nm, 568 nm, 605 and 640 nm for PCzTh–TVCHO, PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT respectively, from which the optical bandgaps were calculated to be 2.46 eV for PCzTh–TVCHO, 2.18 eV for PCzTh–TVCN, 2.05 eV for PCzTh–TVDCN, and 1.94 eV for PCzTh–TVDT. The absorption peak wavelength and optical bandgaps of the copolymer films are also listed in Table 1. The variation in the absorption and bandgap suggests the possibility of tuning the photophysical properties by the simple manipulation of the acceptor strength in the pendant groups.
Electrochemical properties and electronic energy levels
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the conjugated polymers are important parameters in the design of optoelectronic devices, and they can be estimated from the onset oxidation and reduction potentials.40 As shown in Table 1, the onset oxidation potentials (Eoxonset) are 0.85 V for PCzTh–TVCHO, 0.86 V for PCzTh–TVCN, 0.89 V for PCzTh–TVDCN, and 0.88 V vs. Ag/Ag+ for PCzTh–TVDT. The HOMO energy levels of the polymers were estimated according to the following equations:41EHOMO = −e(Eoxonset + 4.71) (eV), where the unit of Eoxonset is V vs. Ag/Ag+. The HOMO levels of PCzTh–TVCHO, PCzTh–TVCN and PCzTh–TVDCN, calculated in this way, are −5.56 eV, −5.57 eV and −5.60 eV and −5.59 eV, respectively, as listed in Table 1. Since we couldn't obtain reliable onset reduction potentials for the four copolymers, the optical band gaps (Eg) of the polymer films (which were calculated from their absorption band edge) were utilized to estimate the LUMO energy levels of the copolymers based on the relation of Eg = ELUMO − EHOMO. The estimated LUMO energy levels of PCzTh–TVCHO, PCzTh–TVCN and PCzTh–TVDCN and PCzTh–TVDT are −3.10 eV, −3.39 eV, −3.55 eV and −3.65 eV, respectively, which are also shown in Table 1. Obviously, the HOMO energy levels of the four copolymers remain almost unchanged, in spite of the presence of different pendant acceptor groups, while their LUMO energy levels decreased after attaching the acceptor cyano and 1,3-diethyl-2-thiobarbituric acid (DT) acceptor groups in the conjugated side chains on the Th units. Regarding that the threshold HOMO level for air stable conjugated polymers being estimated to be −5.2 eV,42,43 lower HOMO levels of the three copolymers with cyano and DT acceptor groups should be beneficial to their chemical stability in ambient conditions. In addition, the deeper HOMO level of the copolymers is desirable for higher open circuit voltage (Voc) of the PSCs with polymers as donor materials because the Voc is usually proportional to the difference between the LUMO level of the acceptor and the HOMO level of the donor.9Theoretical calculations
To provide further insight into the fundamentals of molecular architecture, molecular simulation was carried out for PCzTh–TVCHO, PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT with a chain length of n = 1 at the DFT B3LYP/6-31G(d) level with the Gaussian 03 program package.42 The bulky N-alkyl chain was not included in the calculation to avoid excessive computation demand, since they do not significantly affect the equilibrium geometry and thus the electronic properties. All CC bonds outside the aromatic rings were set to be in trans configuration. As depicted in Fig. 4, the HOMOs for all the four polymers are located on the peripheral thiophene groups and the adjacent carbazole rings, and the computed HOMO values are close to one another for the four polymers. The electron density distributions at LUMO become highly localized near the electron-withdrawing group, and the well-separated distribution of HOMO and LUMO levels indicate that a transition between them can be considered as a charge-transfer transition. The unique electron distribution patterns of PCzTh–TVCN, PCzTh–TVDCN, and PCzTh–TVDT have the advantage of allowing charge separation through sequential transfer of electrons from the main chains to the side chain acceptor groups and then to fullerene acceptor in a bulk-heterojunction PSC. Also, it can be envisioned from the DFT results that LUMO energy levels can be further decreased by appending a stronger pedant acceptor within this molecular architecture, indicative of flexibility of this special main chain donor-side chain acceptor D–A copolymer architecture for tuning the opto-electronic properties of the conjugated polymers.
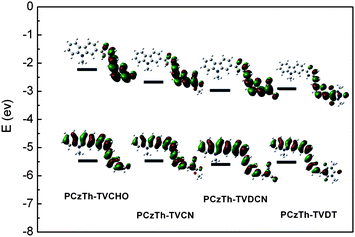 | ||
| Fig. 4 HOMO and LUMO energy levels and the frontier molecular orbital obtained from DFT calculations on PCzTh–TVCHO, PCzTh–TVCN, PCzTh–TVDCN and PCzTh–TVDT with a chain length n = 1 at B3LYP/6-31G* level of theory. | ||
In addition, it can be seen from Fig. 4 and Table 1 that although discrepancies exist between the calculation and experimental results, the trends of variation in the HOMO and LUMO energy levels, as well as the energy gaps, are similar. The presence of pendant acceptor groups with strong electron affinity leads to a decrease in the LUMO energies, but affects the HOMO levels to a much lesser extent. This phenomenon is significantly different from most linear D–A type polymers, whose HOMO levels were elevated a lot when using different acceptors to reduce the band gaps.43
Photovoltaic properties
To balance the absorbance and the charge transporting network of the photoactive layer, the weight ratios of copolymer and PC60BM were varied from 1:1 to 1:4 with the device configuration of ITO/PEDOT: PSS/copolymer: PC60BM/Al. Table 2 clearly shows the dependence of the short circuit current density (Jsc) and power conversion efficiency (PCE) on the blend weight ratios. Also it worth noting that Jsc is strongly affected by the concentration of PC60BM in the blend, while the open circuit voltage (Voc) is much less dependent on the concentration of PC60BM as it is related to the offset between the two components. A higher concentration of PC60BM should favor the formation of the interpenetrated networks of the fullerene acceptor, which in turn favors the effective charge separation and charge transportation. Further increase of the PC60BM concentration over the weight ratio of 1:3 resulted in decrease of the Jsc and PCE values of the PSCs, probably due to the reduction of the amount of polymer donor, which results in the reduction of absorption and hole transporting ability of the active layer.44 Thus a 1:3 weight ratio was chosen as the reference ratio for further optimization.
| Polymers | Ratio (w:w) | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Thickness (nm) |
|---|---|---|---|---|---|---|
| PCzTh–TVCN | 1:1 |
0.77 | 2.26 | 32.0 | 0.56 | 42 |
| 1:2 |
0.83 | 3.16 | 33.9 | 0.89 | 62 | |
| 1:3 |
0.83 | 3.21 | 38.3 | 1.02 | 45 | |
| 1:4 |
0.84 | 3.17 | 34.5 | 0.92 | 93 | |
| PCzTh–TVDCN | 1:1 |
0.78 | 3.40 | 35.3 | 0.93 | 60 |
| 1:2 |
0.96 | 3.66 | 32.8 | 1.15 | 63 | |
| 1:3 |
0.95 | 4.00 | 35.2 | 1.34 | 95 | |
| 1:4 |
0.93 | 3.56 | 33.6 | 1.12 | 78 | |
| PCzTh–TVDT | 1:1 |
0.75 | 2.40 | 27.0 | 0.49 | 57 |
| 1:2 |
0.90 | 3.15 | 28.4 | 0.81 | 64 | |
| 1:3 |
0.94 | 3.70 | 28.7 | 1.00 | 70 | |
| 1:4 |
0.93 | 3.20 | 27.4 | 0.82 | 68 |
In the PSCs, the driving force for charge migration is considered to be the internal electric field produced by the work function difference of the anode and cathode. Therefore, to improve the performance of the devices, Ca/Al electrode with lower work function was used to fabricate the organic solar cell devices. Also to further improve the absorption of the composite film, PC70BM was used as acceptors. Photovoltaic performance characteristics of PSCs are shown in Fig. 5, and summarized in Table 3. Combined with an increased Voc and Jsc, the PSCs give better PCEs.
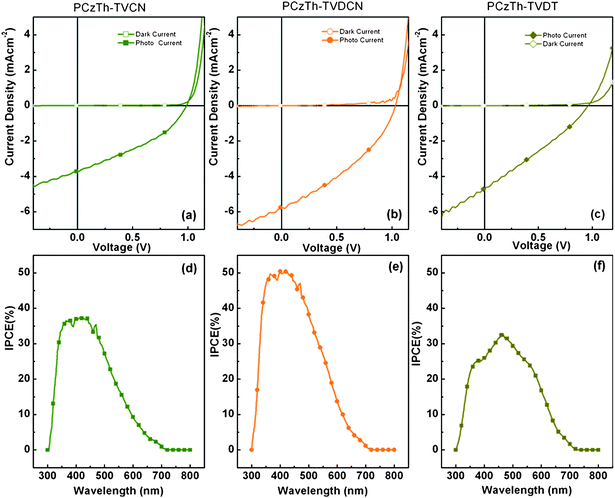 | ||
| Fig. 5 Current density-voltage characteristics of PSCs based on the 1:3 weight ratio composite films of PCzZTH–TVCN:PC70BM (a), PCzTh–TVDCN:PC70BM (b) and PCzTh–TVDT:PC70BM (c). The incident-photon-to-converted-current efficiency (IPCE) spectra of the corresponding devices: PCzTh–TVCN:PC70BM (d), PCzTh–TVDCN:PC70BM (e) and PCzTh–TVDT:PC70BM (f). | ||
:3, w/w)
| Polymers | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Thickness (nm) | Mobility (cm2/VS) |
|---|---|---|---|---|---|---|
| PCzTh–TVCN | 0.99 | 3.72 | 36.1 | 1.33 | 58 | 1.02 × 10−5 |
| PCzTh–TVDCN | 1.03 | 5.75 | 36.5 | 2.16 | 55 | 2.96 × 10−5 |
| PCzTh–TVDT | 0.96 | 4.67 | 29.1 | 1.30 | 63 | 1.16 × 10−7 |
The PSC based on PCzTh–TVCN demonstrates a Jsc of 3.72 mA cm−2, a Voc of 0.99 V, a FF of 0.361, and PCE of 1.33%. The PSC based on PCzTh–TVDCN shows a better photovoltaic performance with JSC of 5.75 mA cm−2, Voc of 1.03 V, FF of 0.365, and PCE of 2.16%. In comparison with PCzTh–TVCN, the better overlap of the absorption spectrum of PCzTh–TVDCN with the solar spectrum and inherent charge separation driving force between backbone donor and pedant cyano group brought by stronger ICT interaction, can account for the increase in JSC, as revealed by incident-photon-to-current efficiency (IPCE) spectra (Fig. 5(d)). Also, better photovoltaic performance of the copolymers could be expected by extending the absorption into longer wavelength by attaching longer planar π-bridge between the donor and acceptors.
The power conversion efficiency obtained for devices based on PCzTh–TVDT is considerably lower than that for devices based on PCzTh–TVDCN, despite the better match between absorption spectra of the former and the solar irradiance spectrum.
The electron deficient nature of the DT group provides wider photo responsive range (Fig. 5(f)), but can also act as the trapping site, prohibiting electron transfer from the main chain to the acceptor, consequently low IPCE value was obtained.
Considering that absorption associated exciton generation is one of the three factors for carrier generation. The other two factors, namely, the diffusion of excitons to the heterojunction and the transport of separated charges to the electrodes, should also be taken account. Thus, to clarify the effect of pedant acceptors on the device performance, the morphology of the composite films was studied by atomic force microscopy (AFM) and charge mobility in blend films was measured by SCLC method.
Morphology and mobility
Hole mobility of the three copolymers was measured by space charge limit current (SCLC) method. PCzTh–TVDCN containing a dicyano modified side chain group, demonstrated an improved hole mobility of 2.96 × 10−5 cm2V−1s−1), over double that of PCzTh–TVCN (1.02 × 10−5 cm2V−1s−1) and two orders higher than that of PCzTh–TVDT (1.16 × 10−7 cm2V−1s−1) (Table 3). Furthermore, the relatively low hole mobility of the polymers partially explains why films thinner than 100 nm need to be employed in all cases.The morphologies of the blend of PC70BM and the three copolymers of PCzTh–TVCN, PCzTh–TVDCN, and PCzTh–TVDT, were investigated by atomic force microscopy (AFM). As shown in Fig. 6, all the blend films demonstrated significant different phase separation state with root-mean-square (rms) roughness of 3.382 nm for PCzTh–TVCN, 0.327 nm for PCzTh–TVDCN, and 0.534 nm for PCzTh–TVDT, indicating different miscibility between the copolymers and PC70BM. From the cross-section profiles, domain size of blend film is estimated to be about 120 nm PCzTh–TVCN, 20 nm for PCzTh–TVDCN and 50 nm for PCzTh–TVDCN; morphology of PCzTh–TVDCN seems much closer to the ideal phase separation scale. For PCzTh–TVCN, the long alkoxy chains can account for substantial phase segregation and high surface roughness.33 The ideal phase separation together with the noticeably higher hole mobility of PCzTh–TVCN facilitates an improved exciton dissociation and carrier collection efficiency, thus leading to an increase in short-circuit current density as well as device efficiency.
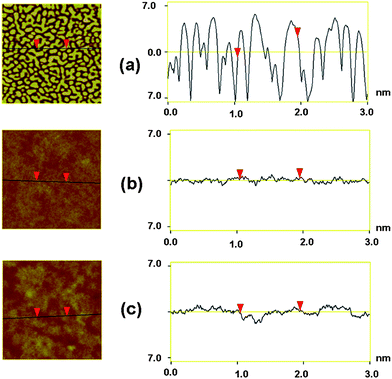 | ||
| Fig. 6
Tapping mode AFM topography images (3 × 3 μm2) of the 1:3 (weight ratio) composite films of (a) PCzTh–TVCN/PC70BM (b) PCzTh–TVDCN/PC70BM (c) PCzTh–TVDT/PC70BM and the corresponding topographical roughness. | ||
4. Conclusion
A conjugated alternating copolymer of carbazole and thiophene bearing the thienylene-vinylene conjugated side chain with aldehyde terminal group, PCzTh–TVCHO, was synthesized via the Suzuki coupling method. Then three copolymers with the side chains containing acceptor groups of mono-cyano (PCzTh–TVCN), di-cyano (PCzTh–TVDCN), and 1,3-diethyl-2-thiobarbituric acid (PCzTh–TVDT), respectively, were synthesized via chemical transformation of the aldehyde group in PCzTh–TVCHO, into respective acceptor groups. The synthesis strategy allows direct comparison of polymers with the same number of conjugation units for elucidating their structure–properties relationship. The copolymers exhibit good solubility in common organic solvents and good thermal stability. Through manipulating the strength of the acceptor groups attached at the conjugated side chain of the copolymer backbone, the energy levels, and thus the photophysical and electronic properties, as well as the absorption spectra of the copolymers were effectively tuned. The chemical transformation approach thus provides a simple and effective way for tuning the optoelectronic properties of the conjugated polymers. Based on comparison and analysis on absorption, morphology, mobility, and photovoltaic property of copolymers, it can be inferred that the dicyano group (DCN) is the proper acceptor in the copolymer with carbazole and thiophene main chain and thienylene-vinylene conjugated side chain. The power conversion efficiency of the PSCs based on the PCzTh–TVDCN/PC70BM (1:3, w/w) reached 2.16% with a high Voc of 1.03 V, under the illumination of AM1.5, 100 mW cm−12. The selection of end groups on side chains is of vital importance involving the D–A character and polarity of the end groups.
Acknowledgements
This work was supported by NSFC (Nos. 20874106, 20821120293 and 50933003), and the Chinese Academy of Sciences. The authors thank Dr Shanghui Ye for the AFM measurements.References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- (a) J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS; (b) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS; (c) A. Facchetti, Chem. Mater., 2011, 23, 733–758 Search PubMed; (d) X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS.
- Y. F. Li and Y. P. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
- (a) Y. J. He, H.-Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS; (b) Y. J. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC; (c) G. J. Zhao, Y. J. He and Y. F. Li, Adv. Mater., 2010, 22, 4355–4358 CrossRef CAS; (d) Y. J. He, G. J. Zhao, B. Peng and Y. F. Li, Adv. Funct. Mater., 2010, 20, 3383–3389 CrossRef CAS.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS.
- O. Inganäs, F. Zhang, K. Tvingstedt, L. M. Andersson, S. Hellström and M. R. Andersson, Adv. Mater., 2010, 22, E100–E116 CrossRef.
- Barry C. Thompson and Jean M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef.
- Z.-G. Zhang, K.-L. Zhang, G. Liu, C.-X. Zhu, K.-G. Neoh and E.-T. Kang, Macromolecules, 2009, 42, 3104–3111 CrossRef CAS.
- H. Zhou, L. Yang, S. C. Price, K. J. Knight and W. You, Angew. Chem., Int. Ed., 2010, 49, 7992–7995 Search PubMed.
- (a) R. Mondal, S. Ko and Z. Bao, J. Mater. Chem., 2010, 20, 10568–10576 RSC; (b) E. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302–8305 CrossRef CAS; (c) E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 Search PubMed; (d) E. Zhou, Q. Wei, S. Yamakawa, Y. Zhang, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2009, 43, 821–826 Search PubMed.
- Y. Zhang, S. K. Hau, H.-L. Yip, Y. Sun, O. Acton and A. K. Y. Jen, Chem. Mater., 2010, 22, 2696–2698 CrossRef CAS.
- Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394–5404 CrossRef CAS.
- J.-S. Wu, Y.-J. Cheng, M. Dubosc, C.-H. Hsieh, C.-Y. Chang and C.-S. Hsu, Chem. Commun., 2010, 46, 3259–3261 RSC.
- J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS.
- L. J. Huo, J. H. Hou, S. Q. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CAS.
- S. C. Price, A. C. Stuart and W. You, Macromolecules, 2010, 43, 4609–4612 CrossRef CAS.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage Jr, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J.-L. Bredas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123–134 RSC.
- E. J. Zhou, J. Cong, S. Yamakawa, Q. Wei, M. Nakamura, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2010, 43, 2873–2879 CrossRef CAS.
- Y. P. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS.
- Z. Li, J. Ding, N. Song, J. Lu and Y. Tao, J. Am. Chem. Soc., 2010, 132, 13160–13161 CrossRef CAS.
- M. J. Zhang, H. J. Fan, X. Guo, Y. J. He, Z.-G. Zhang, J. Min, J. Zhang, G. J. Zhao, X. W. Zhan and Y. F. Li, Macromolecules, 2010, 43, 8714–8717 CrossRef CAS.
- H. Xin, X. Guo, F. S. Kim, G. Ren, M. D. Watson and S. A. Jenekhe, J. Mater. Chem., 2009, 19, 5303–5310 RSC.
- (a) J. H. Hou, L. J. Huo, C. He, C. H. Yang and Y. F. Li, Macromolecules, 2005, 39, 594–603 Search PubMed; (b) J. H. Hou, Z. A. Tan, Y. Yan, Y. J. He, C. H. Yang and Y. F. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS; (c) J. H. Hou, Z. A. Tan, Y. J. He, C. H. Yang and Y. F. Li, Macromolecules, 2006, 39, 4657–4662 CrossRef CAS; (d) E. J. Zhou, Z. A. Tan, L. J. Huo, Y. J. He, C. H. Yang and Y. F. Li, J. Phys. Chem. B, 2006, 110, 26062–26067 CrossRef CAS.
- Y. Wang, E. J. Zhou, Y. Q. Liu, H. Xi, S. Ye, W. Wu, Y. Guo, C.-A. Di, Y. Sun, G. Yu and Y. F. Li, Chem. Mater., 2007, 19, 3361–3363 CrossRef CAS.
- Y. P. Zou, W. Wu, G. Y. Sang, Y. Yang, Y. Q. Liu and Y. F. Li, Macromolecules, 2007, 40, 7231–7237 CrossRef CAS.
- F. Huang, K.-S. Chen, H.-L. Yip, S. K. Hau, O. Acton, Y. Zhang, J. Luo and A. K. Y. Jen, J. Am. Chem. Soc., 2009, 131, 13886–13887 CrossRef CAS.
- (a) C. Duan, W. Cai, F. Huang, J. Zhang, M. Wang, T. Yang, C. Zhong, X. Gong and Y. Cao, Macromolecules, 2010, 43, 5262–5268 CrossRef CAS; (b) C. Duan, K.-S. Chen, F. Huang, H.-L. Yip, S. Liu, J. Zhang, A. K. Y. Jen and Y. Cao, Chem. Mater., 2010, 22, 6444–6452 CrossRef CAS; (c) C. Duan, C. Wang, S. Liu, F. Huang, C. H. W. Choy and Y. Cao, Sci. China Chem., 2011, 54, 685–694 Search PubMed.
- Z.-G. Zhang, Y.-L. Liu, Y. Yang, K. Hou, B. Peng, G. J. Zhao, M. Zhang, X. Guo, E.-T. Kang and Y. F. Li, Macromolecules, 2010, 43, 9376–9383 CrossRef CAS.
- (a) S.-L. Hsu, C.-M. Chen and K.-H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5126–5134 Search PubMed; (b) Y.-J. Cheng, L.-C. Hung, F.-Y. Cao, W.-S. Kao, C.-Y. Chang and C.-S. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1791–1801 Search PubMed; (c) D. Sahu, H. Padhy, D. Patra, J.-h. Huang, C.-w. Chu and H.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5812–5823 Search PubMed.
- H. J. Fan, Z.-G. Zhang, Y. F. Li and X. W. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1462–1470 Search PubMed.
- (a) P.-L. T. Boudreault, S. Beaupré and M. Leclerc, Polym. Chem., 2010, 1, 127–136 RSC; (b) N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS.
- (a) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS; (b) E. Zhou, S. Yamakawa, K. Tajima, C. Yang and K. Hashimoto, Chem. Mater., 2009, 21, 4055–4061 CrossRef CAS; (c) E. Zhou, J. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890–4895 CrossRef CAS.
- R. Qin, W. Li, C. Li, C. Du, C. Veit, H.-F. Schleiermacher, M. Andersson, Z. Bo, Z. Liu, O. Inganäs, U. Wuerfel and F. Zhang, J. Am. Chem. Soc., 2009, 131, 14612–14613 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 130, 732–742.
- R. Pokrop, J. M. Verilhac, A. Gasior, I. Wielgus, M. Zagorska, J. P. Travers and A. Pron, J. Mater. Chem., 2006, 16, 3099–3106 RSC.
- M. Surin, P. Sonar, A. C. Grimsdale, K. Mullen, S. De Feyter, S. Habuchi, S. Sarzi, E. Braeken, A. V. Heyen, M. Van der Auweraer, F. C. De Schryver, M. Cavallini, J. F. Moulin, F. Biscarini, C. Femoni, L. Roberto and P. Leclere, J. Mater. Chem., 2007, 17, 728–735 RSC.
- Y. F. Li, Y. Cao, J. Gao, D. L. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243–248 CrossRef CAS.
- Q. J. Sun, H. Q. Wang, C. H. Yang and Y. F. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- R.E. Gaussian 03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, J. Vreven, T. K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- J. H. Hou, M.-H. Park, S. Q. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- C. He, Q. G. He, X. Yang, G. Wu, C. H. Yang, F. L. Bai, Z. G. Shuai, L. Wang and Y. F. Li, J. Phys. Chem. C, 2007, 111, 8661–8666 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |