Synthesis and application of new CO2-soluble vinyl pivalate hydrocarbon stabilisers viaRAFT polymerisation†
Natasha A.
Birkin
,
Nicholas J.
Arrowsmith
,
Eun Ju
Park
,
Alexandre P.
Richez
and
Steven M.
Howdle
*
School of Chemistry, University of Nottingham, Nottingham, UK NG7 2RD. E-mail: steve.howdle@nottingham.ac.uk; Tel: +44 (0)1159513486
First published on 20th April 2011
Abstract
We describe the synthesis of novel, highly CO2-soluble stabilisers that overcome the current limitation requiring use of fluorinated or siloxane based materials for synthesis and processing. A series of homopolymers and copolymers with targeted molecular weight (∼10 kg mol−1) of vinyl pivalate and vinyl acetate were prepared by xanthate mediated RAFT polymerisation and a range of comonomer ratios have been characterised by GPC, NMR and DSC. In addition, detailed phase behaviour studies using a variable volume view cell were used to determine the relative solubility in scCO2. The stabilisers were applied to scCO2 dispersion polymerisations to determine their efficacy and SEM was primarily used to gauge the stabiliser effect upon the microparticle morphology of the polymer product.
Introduction
Supercritical carbon dioxide (scCO2) is an attractive alternative to conventional solvents for dispersion polymerisation because it is environmentally acceptable, inexpensive and leaves no solvent residues.1–3 Moreover, proposed global carbon capture strategies indicate that scCO2 will be a widely available and inexpensive solvent for widespread future use. A perceived major drawback to the use of scCO2 is the poor solubility of high molecular weight materials.4–8 But, this can be a distinct advantage for heterogeneous polymerisations. The major hurdle to overcome for any heterogeneous processing is the need for highly soluble dispersants or stabilisers that are commercially viable and environmentally acceptable.Up till now, only fluorinated or silicone based materials have shown real promise, because of their very strong interactions with CO2. Over the last two decades a range of fluorinated stabilisers, such as Krytox 157-FSL and poly(1,1-dihydroperfluorooctyl acrylate) (PFOA), were successfully used in dispersion polymerisations in scCO2.9–13 More recently silicones, such as poly(dimethylsiloxane) (PDMS), have also shown promise as successful stabilisers for methyl methacrylate (MMA) polymerisations in scCO2.14,15 To date a range of fluorinated and silicone homopolymer, block copolymer and graft copolymer structures have been employed in dispersion polymerisations.3,13,16–20 However, using fluorocarbons and silicones has disadvantages, namely their potential toxicity and high cost. The lack of inexpensive alternative stabilisers is perhaps one of the key reasons why scCO2 processes have not been widely commercially adopted. Recently there has been an increasing drive to discover new CO2 soluble hydrocarbon materials for use in scCO2—where the term hydrocarbon implies that the polymers are silicone and fluorine free.
Beckman and co-workers developed poly(ether-carbonate) copolymers which were able to act as efficient, non-fluorous CO2-philes and readily dissolved at low CO2 pressures.21 Further research on polyethers identified that the presence of an accessible ether oxygen enhanced the CO2-solubility.22Sugar acetate structures have also been proposed as potential CO2-philes.23 There has been much research on the observed high solubility of hydrocarbon polymers such as poly(vinyl acetate) (PVAc)24,25 which contain carbonyl and acetate groups.26–28 The carbonyl group is proposed to be involved in favourable interactions with CO2; the electron-donating oxygen group of the carbonyl promoting Lewis acid–base interactions with the carbon of CO2.29,30
We recently reported the synthesis and application of poly(vinyl alkylate) stabilisers based upon vinyl acetate and vinyl butyrate homopolymers and random copolymers which acted as CO2-soluble hydrocarbon surfactants.31 Here we report significant further advances and the development of a viable, highly scCO2 soluble range of hydrocarbon stabilisers.
Vinyl pivalate (VPi) is a monomer with a structure similar to that of vinyl acetate, with a tertiary butyl group replacing the methyl group of vinyl acetate (Fig. 1). VPi is often used in the preparation of PVA, and microphase separated block copolymers of PVAc and PVPi have also recently been synthesised using the MADIX process.32,33
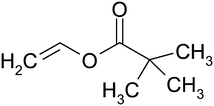 | ||
| Fig. 1 Vinyl pivalate monomer structure. | ||
Our results show that poly(vinyl pivalate) (PVPi) possesses significant solubility in scCO2, comparable with PVAc, at molecular weights typically ∼10 kg mol−1. The presence of a bulky substituent within such materials should disrupt the regularity in the chain packing of polymer chains and result in increased free volume between the chains, lowering the cohesive energy density and improving solubility.5,34,35 In the case of VPi, the additional tertiary butyl group should act to increase the solubility compared to VAc. In this paper we will investigate the effect of VPi upon copolymer solubility in scCO2 and also target the development of novel hydrocarbon surfactants for dispersion polymerisation in scCO2.
Experimental section
Materials
Monomers vinyl acetate (VAc) (99%) and vinyl pivalate (VPi) (99%) were purchased from Sigma Aldrich. N-Vinyl pyrrolidone (NVP) (97%, 0.001% N,N′-di-sec-butyl-p-phenylenediamine inhibitor) was obtained from Fluka. All monomers were stored at 3–4 °C and purified prior to use by passing through a column of activated aluminium oxide, and subsequently degassing via 3 freeze–pump–thaw cycles. The initiator 2,2′-azobis(isobutyronitrile) (AIBN) was obtained from Acros and purified by recrystallisation twice from cold methanol. Initiator 2,2′-azobis (4-methoxy-2,4-dimethylvaleronitrile) (V-70) (WAKO, 95%) was used as received. Dry CO2 (99.99%) and nitrogen (99.99%) were purchased from BOC.Synthesis of hydrocarbon stabilisers
All stabilisers were prepared in a controlled manner viaRAFT polymerisation, using the RAFT agent S-(1-ethoxycarbonylethyl) O-ethyl xanthate (X1), synthesised as previously reported.36Typical 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 random copolymer synthesis: vinyl acetate (3.73 g, 3.6 × 10−2 mol), vinyl pivalate (4.67 g, 3.6 × 10−2 mol), xanthate X1 (0.13 g, 5.7 × 10−4 mol), AIBN (0.009 g, 5.7 × 10−5 mol) and dry toluene (5 ml) were added to a 50 ml round bottomed flask equipped with a stirrer bar and a 3-way stop cock. The flask was then thoroughly degassed using a 3-way freeze–pump–thaw cycle. The flask was immersed in an oil bath at 65 °C for 48 h. The polymer product was purified viaprecipitation into a mixture of ice cold methanol:water (4:1). The mixture was filtered and the solid product vacuum dried. The ratio of the RAFT agent (R) to the initiator (I) concentration used was [R]:[I] = 1:0.1. Homopolymerisations were carried out in bulk, whilst copolymers were synthesised via solution polymerisation in toluene (Fig. 2).
:50 random copolymer synthesis: vinyl acetate (3.73 g, 3.6 × 10−2 mol), vinyl pivalate (4.67 g, 3.6 × 10−2 mol), xanthate X1 (0.13 g, 5.7 × 10−4 mol), AIBN (0.009 g, 5.7 × 10−5 mol) and dry toluene (5 ml) were added to a 50 ml round bottomed flask equipped with a stirrer bar and a 3-way stop cock. The flask was then thoroughly degassed using a 3-way freeze–pump–thaw cycle. The flask was immersed in an oil bath at 65 °C for 48 h. The polymer product was purified viaprecipitation into a mixture of ice cold methanol:water (4:1). The mixture was filtered and the solid product vacuum dried. The ratio of the RAFT agent (R) to the initiator (I) concentration used was [R]:[I] = 1:0.1. Homopolymerisations were carried out in bulk, whilst copolymers were synthesised via solution polymerisation in toluene (Fig. 2).
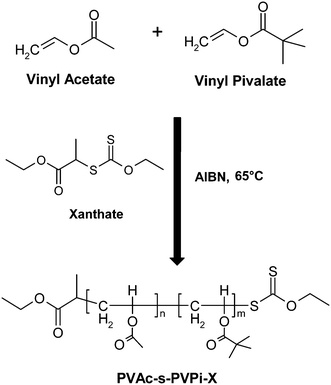 | ||
| Fig. 2 Synthesis of xanthate terminated PVAc-s-PVPi-X statistical copolymer. | ||
Hydraulic variable volume view cell
Cloud point curves were obtained using a hydraulic variable volume view cell.37 A known amount of polymer was added to the view cell body and a high-pressure stainless steel bomb was used to deliver CO2 to the cell. Stirring was maintained using a magnetic stirrer bar. The system was heated to the desired temperature and allowed to equilibrate. Cloud point measurements were determined by gradually retracting the piston and decreasing the volume within the view cell, until the polymer sample precipitated and the three point light sources at the back of the view cell could no longer be seen through the solution (see ESI†). All cloud points were repeated three times. Cloud point measurements were recorded from 35–75 °C.High pressure polymer synthesis
Dispersion polymerisations were performed in a 60 ml autoclave equipped with a magnetically driven overhead stirrer. 1-Vinyl-2-pyrrolidone (NVP) was used as the monomer (8.32 g, 0.072 mol). 5 weight% of the stabiliser material was used with respect to the monomer, and 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) was employed as an initiator in the ratio [S]:[I] = 1:10, where [S] is the stabiliser and [I] the initiator. The reaction proceeded for 48 hours at 35 °C and 4000 psi (27.6 MPa). The autoclave was then cooled via immersion in an acetone and dry ice bath, and vented slowly.
Characterisation
The molecular weight and the polydispersity of the hydrocarbon stabiliser samples were determined using Gel Permeation Chromatography (PL-GPC 120, Polymer Labs) with differential refractometer detection. THF was employed as an eluent, with 2 columns (30 cm, PolarGel-M) in series calibrated against polystyrene standards. N,N′-Dimethylformamide with 0.01 M lithium bromide was employed as the eluent for characterisation of poly(vinyl pyrrolidone) (PVP) samples, with calibration using PMMA standards. Differential Scanning Calorimetry (DSC) analysis was obtained using a DSC Q2000 for thermal analysis. Measurements were run at 10 °C min−1 with a nitrogen flow rate of 50 ml min−1, and a temperature range between 0 °C and 100 °C. Determination of the composition ratios for the statistical copolymers, and the monomer conversion for all polymers, was calculated from the relevant peaks of the 1H NMR spectra recorded using a Bruker DPX-300 Spectrometer in CDCl3. SEM analysis of PVP products was carried out using a JEOL 6060LV variable pressure scanning electron microscope. Samples were prepared using a Balzers SCD 030 gold sputter coater. Mean particle diameter (Dn, μm) was determined by measuring the diameter of ca. 100 particles. The coefficient of variance (Cv), an indication of particle size distribution, was determined using the equation Cv = (σ/Dn) × 100, where σ corresponds to the standard deviation of the particle diameter (μm).Results and discussion
Vinyl pivalate homopolymers
Poly(vinyl pivalate) (PVPi) stabilisers of varying targeted molecular weights were synthesised as described using the xanthate RAFT agent (Table 1). All of the xanthate terminated homopolymers were obtained as a white, free flowing powder and characterised using 1H NMR and GPC.| Entry |
M:R:I |
M n(th.) /kg mol−1 | M n(expt.) b/kg mol−1 | PDIb | Conv.c (%) |
|---|---|---|---|---|---|
| a Polymerisation conditions: bulk polymerisation at 65 °C for 24 hours. b Experimental Mn and the PDI obtained from GPC-RI detector using PS standards. c Conversion determined from 1H NMR in CDCl3. | |||||
| 1 | 38:1:0.1 |
5.0 | 5.0 | 1.4 | 80 |
| 2 | 60:1:0.1 |
7.9 | 7.8 | 1.4 | 85 |
| 3 | 77:1:0.1 |
10.0 | 10.0 | 1.3 | 85 |
| 4 | 109:1:0.1 |
14.2 | 14.3 | 1.4 | 79 |
| 5 | 139:1:0.1 |
18.2 | 17.5 | 1.5 | 83 |
| 6 | 160:1:0.1 |
20.8 | 20.5 | 1.5 | 90 |
Monomer conversion was calculated from comparison of monomer and polymer peaks of the1H NMR at 4.5–4.6 and 4.8–5.0 ppm respectively (ESI†). Molecular weights achieved were close to the theoretical Mn, and PDI values were acceptable (<1.5), indicating a controlled polymerisation. Overall, PDI appeared to increase with increasing molecular weight.
Copolymers of vinyl pivalate and vinyl acetate
Statistical copolymers of vinyl pivalate and vinyl acetate were synthesised using the xanthate S-(1-ethoxycarbonylethyl) O-ethyl xanthate. Copolymerisations were carried out in the same manner as vinyl pivalate homopolymerisations to yield PVAc-s-PVPi-X (Table 2).| Entry | M n(th.) | M n(expt.) b | PDIb | Feed ratio | Expt. ratioc | Conv.c (%) | T g d/°C |
|---|---|---|---|---|---|---|---|
|
a
Polymerisation conditions: solution polymerisation in dry toluene (5 ml) at 65 °C for 48 hours. Ratios correspond to PVAc:PVPi composition.
b Experimental Mn and PDI obtained from GPC-RI detector using PS standards.
c Conversion determined from 1H NMR in CDCl3.
d
T
g values obtained from DSC analysis.
|
|||||||
| 1 | 14.0 | 8.8 | 1.6 | 75:25 |
72:28 |
66 | 32.0 |
| 2 | 20.0 | 15.6 | 1.4 | 50:50 |
47:53 |
80 | 56.3 |
| 3 | 14.0 | 9.4 | 1.5 | 50:50 |
44:56 |
65 | 47.4 |
| 4 | 14.0 | 10.3 | 1.4 | 25:75 |
24:76 |
63 | 52.3 |
| 5 | 16.0 | 10.6 | 1.5 | 10:90 |
16:84 |
85 | 57.8 |
| 6 | 15.0 | 8.9 | 1.5 | 8:92 |
10:90 |
82 | 64.6 |
| 7 | 21.0 | 12.8 | 1.6 | 8:92 |
8:92 |
83 | 66.9 |
| 8 | 15.0 | 9.6 | 1.5 | 5:95 |
6:94 |
90 | 65.6 |
The monomer conversion and copolymer composition were determined from 1H NMR (ESI†). The final copolymer composition varied little from the initial monomer feed ratio, an observation which is attributed in part to the extended reaction time. When shorter reaction times are employed, the situation is different. Bulk polymerisations carried out for ∼3 h with equal proportions of VAc and VPi in the feed ratio resulted in a copolymer with ∼40:60 ratio of PVAc:PVPi, indicating the polymerisation time will affect the final compostion. Copolymers with both a range of molecular weights and ratios were targeted in order to study the effects on CO2-solubility. Molecular weights did not reach theoretical values but, target molecular weights could be manipulated successfully to obtain the desired product.
VAc is known to be difficult to polymerize; molecular weights are difficult to predict and the PDI of VAc products tends to be high as the VAc lacks a conjugating substituent, making its propagating radical less stable and encouraging chain transfer and termination.38 PDI values were <1.5 in the majority of cases, indicating controlled polymerisation. The broadest PDI (1.6) was exhibited by the copolymer with the largest proportion of VAc moieties in the structure (entry 1). Although relatively broad, the PDI values obtained are not uncommon for MADIX polymerisation. Stenzel et al. have previously highlighted the requirements for an effective RAFT agent for VAc polymerisations.39 The RAFT agent employed in this work was selected to provide an intermediate radical which would fragment quickly due to a relative instability, provided by an increased electron density at the radical centre, leading to an effective reversible addition fragmentation reaction of VAc. However, the PDI results indicate an even more appropriate RAFT agent may be required. It is also important to note there will be some error in the PDI due to calibration against PS standards and a difference in the hydrodynamic volumes of PVPi/PVAc and PS. Absolute molecular weights were also estimated from comparison of the polymer backbone protons and those of the terminal xanthate in the 1H NMR. Molecular weights determined via1H NMR correlated well with the results from GPC analysis (ESI†).
Differential scanning calorimetry (DSC) was used to understand the effect of the PVAc:PVPi composition on the thermal properties of the copolymers. The glass transition temperature (Tg) was determined by analysis of the large endothermic transition in the second and third heating/cooling cycle. PVAc and PVPi homopolymers are reported to have Tg values of 29 °C and 86 °C respectively. As expected, when analysing the copolymers, those with a larger proportion of VPi had a Tg closer to that of PVPi homopolymer, reflecting the decreased flexibility conferred by VPi.
Studies on homopolymer molecular weight in scCO2 + 15 wt% NVP
A series of phase behaviour studies were carried out over a range of molecular weights to determine the solubility properties of the PVPi materials in scCO2 with 15 wt% NVP monomer in order to mimic initial dispersion polymerisation reaction conditions (Table 1).The monomer acts as a co-solvent, promoting solubility of the hydrocarbon surfactants and enabling phase behaviour to be studied. Without the presence of NVP, the cloud point pressures are much higher and could not be measured within the pressure constraints of the view cell. The homopolymers behaved as expected with an increase in molecular weight and subsequent chain length leading to a corresponding decrease in solubility and a higher cloud point pressure.35,40
One of the key points to note is the significant impact of molecular weight on solubility. At 5k and 7.8k the cloud points are significantly lower and there is a marked shift to the 10.0k stabiliser (Fig. 3), whilst the difference between other higher molecular weight stabilisers is less marked. Solubility is important, but must also be balanced against the required steric effect of the chain length in any stabiliser.
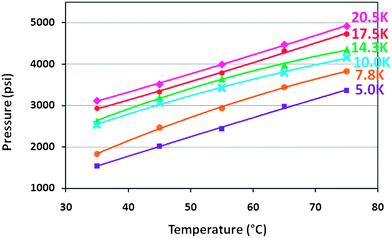 | ||
| Fig. 3 Cloud point curves of PVPi-X homopolymers in CO2. Measurements taken with 15 wt% NVP w.r.t. CO2 and 5 wt% homopolymer w.r.t. monomer and clearly show the increase of the cloud point pressure with the molecular weight. | ||
PVPi was also found to be significantly more soluble than the corresponding PVAc homopolymer of the same molecular weight (Fig. 4). The difference of ∼800 psi indicates that the tertiary butyl group of the VPi moiety strongly influences the solubility. It is also true that the chain lengths will be shorter, but if a comparison is made of homopolymers with similar degree of polymerisation (DP) we find that even a higher molecular weight PVPi-X of 14.3k with similar DP of 110 has a cloud point that is still ∼700 psi lower than the corresponding PVAc of the same backbone length (Table 3, compare entries 1, 2 and 3).
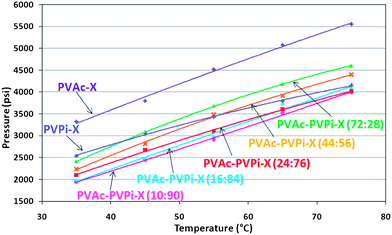 | ||
| Fig. 4 Cloud point curves of PVAc-PVPi-X random copolymers Mn ≈ 10k in CO2, with 15 wt% NVP w.r.t. CO2 and 5 wt% homopolymer w.r.t. monomer. The data clearly show that addition of VPi enhances the solubility. | ||
| Polymer | M n(expt.) a/kg mol−1 | PDIa | Ratio (PVAc:PVPi)b |
DP c | Cloud pointd/psi | T g e/°C |
|---|---|---|---|---|---|---|
| a Experimental Mn and PDI obtained from GPC-RI detector using PS standards. b Ratio determined from 1H NMR in CDCl3. c The degree of polymerisation calculated using Mn and ratio. d Cloud point at 35 °C determined using variable volume view cell (1000 psi = 6.9 MPa). Measurement at 35 °C is given. e T g values obtained from DSC analysis. | ||||||
| PVAc-X | 9.6 | 1.4 | — | 109 | 3321 | 22.9 |
| PVPi-X | 10.0 | 1.3 | — | 76 | 2534 | 66.7 |
| 14.3 | 1.4 | — | 110 | 2625 | 69.7 | |
| PVAc-s-PVPi-X | 8.8 | 1.6 | 72:28 |
88 | 2421 | 32.0 |
| 9.4 | 1.5 | 44:56 |
84 | 2236 | 47.4 | |
| 10.3 | 1.4 | 24:76 |
75 | 2099 | 52.3 | |
| 10.6 | 1.5 | 16:84 |
85 | 1978 | 57.8 | |
| 8.9 | 1.5 | 10:90 |
70 | 1941 | 64.6 | |
| 9.6 | 1.5 | 6:94 |
75 | 2060 | 65.6 |
Studies on copolymer solubility in scCO2 + 15 wt% NVP
Phase behaviour of the random copolymers was carried out to compare the effect of varying composition whilst maintaining a molecular weight of ∼10 kg mol−1 to provide a suitable balance between solubility and stabilising ability (Table 3).The hydrocarbon copolymers were found to possess enhanced solubility with respect to both PVAc and PVPi homopolymers. As might be expected, increasing the proportion of PVPi in the copolymer resulted in a lowering of the cloud point values, and an increased solubility (Fig. 4). Surprisingly, a small proportion of PVAc within the polymer significantly enhances the solubility compared to that of the PVPi homopolymer alone. Whilst VPi residues improve solubility through a larger free volume and decreased polymer–polymer interactions, it is also clear that a small proportion of VAc within the structure introduces a certain degree of flexibility to the otherwise rigid polymer. This flexibility is postulated to further improve the solubility by enabling the polymers to interact more freely with CO2, enhancing the entropy of mixing in the solvent.41 Both residues also possess acetate groups favourable for interaction with CO2. Somewhat counter-intuitively this means that the PVPi copolymers with a higher Tg actually have greater solubility in scCO2 than those with a low Tg.
These solubility studies show great promise and represent a significant step towards inexpensive hydrocarbon stabilisers for application in scCO2.
Effect of copolymer molecular weight on solubility in scCO2 + NVP
The effect of varying molecular weight of the PVAc-s-PVPi-X copolymers on the phase behaviour in scCO2 + NVP co-solvent was investigated. Initially higher molecular weight copolymers with 10:90 and 50:50 feed ratio were targeted.
As expected, the molecular weight trend is the same as observed for the PVPi homopolymers. As the molecular weight increases, the solubility in scCO2 decreases, which is reflected in an increase in the cloud point pressure for each of the stabilisers. As the chain length is shortened, dissolution of the stabilisers in scCO2 occurs more easily (Fig. 5). The 8.9k copolymer has a cloud point of 1940 psi at 35 °C, whilst the corresponding 12.8k copolymer has a cloud point of 2452 psi.
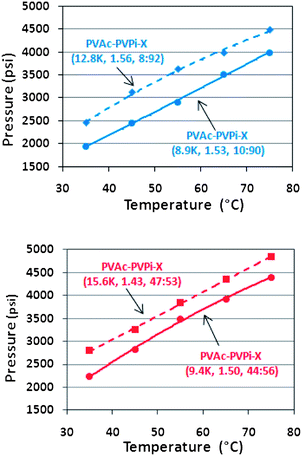 | ||
| Fig. 5 Cloud point curves of copolymers of varying molecular weight at ratios (a) ∼10:90 and (b) ∼50:50. Higher molecular weight copolymers of the same ratio clearly result in a higher cloud point and reduced CO2-solubility, whilst an increased proportion of PVPi also improves the solubility. | ||
This highlights the significant effect molecular weight of the stabiliser will exert on the phase behaviour in scCO2. The same effect was also observed when increasing the PVPi content and considering copolymers with ∼50:50 PVAc:PVPi.
Dispersion polymerisation in scCO2
Dispersion polymerisations in scCO2 were carried out using the NVP monomer (N-vinyl pyrrolidone) in order to understand the stabilising ability of the synthesised poly(vinyl alkylate) materials (Table 4).| Stabiliser | PVP dispersion product | ||||||
|---|---|---|---|---|---|---|---|
| Composition | M n, PDI, ratiob | Cloud pointc/psi | M w d/kg mol−1 | PDId | D n e/μm | C V f (%) | Yieldg (%) |
| a Polymerisation conditions: scCO2 polymerisation at 35 °C for 48 hours with V-70 initiator and 5 wt% of stabiliser. b Stabiliser Mn and PDI determined via GPC-RI with THF eluent and PS standards, ratio determined from 1H NMR. c Stabiliser cloud point determined using a variable volume view cell. d Experimental Mw and PDI obtained from GPC-RI detector in N,N′-dimethylformamide with 0.01 M lithium bromide using PMMA standards. e Mean particle diameter as determined from sampling of ∼100 particles of a typical SEM image. f Coefficient of variance as determined by the equation Cv = (σ/Dn) × 100. g Yield determined gravimetrically. | |||||||
| PVPi-X | 10k, 1.33 | 2534 | 169 | 5.4 | 2.2 | 41.2 | 94 |
| 14.3k, 1.41 | 2625 | 177 | 7.3 | 1.9 | 26.3 | 88 | |
| PVAc-PVPi-X |
9.6k, 1.46, 6:94 |
2060 | 260 | 7.7 | 2.8 | 26.9 | 84 |
|
10.6k, 1.49, 16:84 |
1978 | 169 | 7.3 | 2.4 | 22.6 | 86 | |
|
10.3k, 1.44, 24:76 |
2096 | 149 | 6.8 | 1.7 | 28.0 | 90 | |
|
9.4k, 1.50, 44:56 |
2236 | 139 | 6.5 | 1.4 | 30.2 | 86 | |
|
8.8k, 1.60, 72:28 |
2421 | 133 | 6.1 | 1.3 | 31.3 | 82 | |
GPC analysis of the NVP samples obtained from the polymerisations in scCO2 showed molecular weights in the range of 133–260 kg mol−1. PDI was broad for a typical free radical polymerisation (5.4–7.7), but these results are consistent with those of PVP dispersions in scCO2 by Berger et al., where Mw = 250 kg mol−1 and PDI = 9.7. The unusually high polydispersity was attributed by these authors to “inhomogeneous, non-stationary reaction conditions, and a high radical transfer rate to monomer”.42
All of the polymerisations using the PVPi-based stabilisers exhibited high conversion and the poly(vinyl pyrrolidone) (PVP) sample was obtained as a fine, white, free-flowing powder after venting of the CO2. Particle morphologies were analysed by SEM.
Variation of stabiliser composition
 | ||
| Fig. 6
SEM analysis of the effect on the particle morphology with variation of the copolymer composition at 5 wt% stabiliser loading w.r.t. monomer: (a) PVPi 10.0 K; (b) PVPi 14.3 K; (c) PVAc-PVPi 6:94; (d) PVAc-PVPi 16:84; (e) PVAc-PVPi 24:76; (f) PVAc-PVPi 44:56. Note there is a clear decrease in the particle size at increased VAc ratio (Table 4). | ||
As the molecular weight of the stabiliser was increased to 14.3 K, distinct, spherical particle morphology was observed, indicating the increasing molecular weight was of sufficient length to give adequate steric stabilisation. Stabilisers prepared with even lower DP were previously found to be more soluble in CO2 (Fig. 3) but were not as effective, presumably because they do not possess a sufficient chain length to provide steric stabilisation.
Variation in the copolymer composition was observed to have an effect on the particle morphology. Stabilisers with a very low proportion of VAc residues resulted in particles which were spherical but irregular, and the surfaces distorted. When the composition was adjusted, and 44:56 PVAc:PVPi stabiliser was employed, a decrease in the size of the particles was observed. It appears that particles are smaller and more spherical as the PVAc content incorporated into the stabiliser is increased. The mean particle diameter highlights the differences between the stabilisers with varying compositions, in which as the proportion of VAc increases the particle size is reduced (Table 4). The particle size distribution remains largely similar for the range of compositions, although is observed to be broader for PVPi-X (10.0 K).
Coupled with the improved solubility of copolymers in comparison to homopolymers highlighted in the phase behaviour studies, it is evident that copolymers are more suitable as stabilisers for PVP. SEM images suggest that although stabilisers with a lower VAc content in their composition are most soluble in CO2, these stabilisers also result in increased particle size, suggesting that solubility is not the sole factor to consider when determining the most suitable surfactant for dispersions.
Effect of stabiliser concentration
A study on the effect of the weight percentage of stabiliser employed in scCO2 reactions was carried out. The stabiliser used for this study was PVAc-PVPi-X (9.4 K, 1.50, 44:56), which we have established is known to stabilise successfully at 5 wt% (Fig. 6f).
All weight percentages of the stabiliser gave a white, free flowing powder at a high yield, but the SEM data clearly show that at a low level of 2.0 wt%, only highly distorted near-spherical structures of >10 μm were produced (Fig. 7a) indicating there is insufficient stabiliser present to obtain well defined particle morphology. At higher levels, spherical particles were obtained up to 15 wt% with average diameter <1 μm in size (Fig. 7b–f). As observed in similar studies of surfactant concentration in dispersion systems, increasing concentrations of stabiliser result in a greater surface area of polymer potentially being coordinated to the stabiliser, and lead to a reduction in the particle size of the final product.43,44
 | ||
| Fig. 7 Variation of the stabiliser level: (a) 2.0 wt%, (b) 5 wt%, (c) 7.5wt % at 2000 magnification, (d) 7.5 wt% at 5000 magnification, (e) 15 wt% at 2000 magnification and (f) 15 wt% at 5000 magnification. Particles are well defined and spherical at 5 wt%, and there is a reduction in the particle size as the wt% is increased. | ||
The “gold standard” for stabilisers or dispersants in scCO2 has always been highly soluble fluorinated stabilisers such as Krytox 157-FSL and PFOA. These stabilisers allow for the synthesis of highly spherical, micron-sized particles with high conversion of monomer to polymer, and have been reported to stabilise dispersion polymerisations of PMMA at stabiliser concentrations as low as 0.1 and 0.24 wt% respectively.45,46
However, when converted to molar concentrations it becomes clear that the poly(vinyl alkylates) are performing at a very promising level. For example at 5 wt% (4.5 × 10−5 mol) the poly(vinyl alkylates) are approaching equivalence with the fluorinated Krytox (1 wt%; 4.0 × 10−5 mol), though they do not quite match the extremely low concentrations reported for PFOA.46 Moreover, the poly(vinyl alkylates) are performing as effectively as the more common methacrylate terminated silicone (10 kg mol−1) (5 wt%; 4.0 × 10−5 mol).15 Clearly these findings demonstrate that inexpensive and environmentally acceptable hydrocarbon stabilisers for use in scCO2 are now within reach and could potentially overcome the reliance upon fluorinated or silicone based polymers that have proven to be one of the major barriers to wider commercialisation (ESI†).
Conclusions
We have demonstrated that poly(vinyl alkylate) based stabilisers with high solubility can be successfully synthesised using xanthate mediated RAFT/MADIX polymerisation. Phase behaviour studies have highlighted the significantly improved solubility of both PVPi homopolymers and copolymers as stabilisers. Copolymers comprised of VAc and VPi moieties are significantly more soluble, and increased VPi gives improved solubility. Counter-intuitively, the copolymers with higher Tg are more soluble in scCO2.It has been established that the PVPi based hydrocarbon stabilisers are highly effective for NVP polymerisations, resulting in fine, powder products with high yield and controlled particle morphology. Adjustment of the composition ratio of the stabiliser results in variations of particle morphology, with micron-sized spherical particles. There is a compromise between CO2-solubility and steric stabilising ability, but ∼25:75 to 50:50 ratio of PVAc-PVPi-X stabilisers appear to be most suitable for dispersion polymerisation. These results highlight a route to novel hydrocarbon stabilisers that are inexpensive, highly CO2-soluble and may be applied very successfully in dispersion polymerisations. Future work will exploit these PVPi-based stabilisers, and expand their use in dispersion to other monomer systems, and in development of other surfactant applications.
Acknowledgements
We acknowledge the financial support of the University of Nottingham and EPSRC (EP/F000103) (NAB/APMR/NJA) and also the support of the Korean Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2007-357-D00069) (EJP). We thank also Mr. M. Guyler, Mr. P. Fields and Mr. R. Wilson for invaluable technical support.References
- M. Okubo, Polymer Particles, Springer Publishing, 2005 Search PubMed.
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207–234 RSC.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–563 CrossRef CAS.
- T. Sato and R. Ruch, Stabilisation of Colloidal Dispersions by Polymer Adsorption, Marcel Dekker Inc., 1980 Search PubMed.
- M. L. O'Neill, Q. Cao, R. Fang, K. P. Johnston, S. P. Wilkinson, C. D. Smith, J. L. Kerschner and S. H. Jureller, Ind. Eng. Chem. Res., 1998, 37, 3067–3079 CrossRef CAS.
- R. B. Gupta and J. Shim, Solubility in Supercritical Carbon Dioxide, CRC Press, 2006 Search PubMed.
- S. E. Conway, H. S. Byun, M. A. McHugh, J. D. Wang and F. S. Mandel, J. Appl. Polym. Sci., 2001, 80, 1155–1161 CrossRef CAS.
- M. Lora and M. A. McHugh, Fluid Phase Equilib., 1999, 157, 285–297 CrossRef CAS.
- T. Meyer and J. Keurentjez, Handbook of Polymer Reaction Engineering, Wiley-VCH, 2005 Search PubMed.
- W. H. Tuminello, G. T. Dee and M. A. McHugh, Macromolecules, 1995, 28, 1506–1510 CrossRef CAS.
- J. M. Desimone, E. E. Maury, Y. Z. Menceloglu, J. B. McClain, T. J. Romack and J. R. Combes, Science, 1994, 265, 356–359 CrossRef CAS.
- P. Christian, M. R. Giles, R. M. T. Griffiths, D. J. Irvine, R. C. Major and S. M. Howdle, Macromolecules, 2000, 33, 9222–9227 CrossRef CAS.
- H. M. Woods, M. Silva, C. Nouvel, K. M. Shakesheff and S. M. Howdle, J. Mater. Chem., 2004, 14, 1663–1678 RSC.
- K. A. Shaffer, T. A. Jones, D. A. Canelas and J. A. DeSimone, Macromolecules, 1996, 29, 2704–2706 CrossRef CAS.
- M. R. Giles, J. N. Hay, S. M. Howdle and R. J. Winder, Polymer, 2000, 41, 6715–6721 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- H. S. Ganapathy, H. S. Hwang, M. Y. Lee, Y. T. Jeong, Y. S. Gal and K. T. Lim, J. Mater. Sci., 2008, 43, 2300–2306 CrossRef CAS.
- H. Yuvaraj, H. S. Hwang, W. S. Kim, H. G. Kim, E. D. Jeong and K. T. Lim, Eur. Polym. J., 2008, 44, 2253–2261 CrossRef CAS.
- E. Yoshida, Colloid Polym. Sci., 2008, 286, 351–355 CrossRef CAS.
- Z. Ma and P. Lacroix-Desmazes, Polymer, 2004, 45, 6789–6797 CrossRef CAS.
- T. Sarbu, T. Styranec and E. J. Beckman, Nature, 2000, 405, 165–168 CrossRef CAS.
- C. Drohmann and E. J. Beckman, J. Supercrit. Fluids, 2002, 22, 103–110 CrossRef CAS.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 7274–7275 CrossRef CAS.
- F. Reindfleisch, T. P. DiNoia and M. A. McHugh, J. Phys. Chem., 1996, 100, 15581–15587 CrossRef CAS.
- Z. Shen, M. A. McHugh, J. Xu, J. Belardi, S. Kilic, A. Mesiano, S. Bane, C. Karnikas, E. Beckman and R. Enick, Polymer, 2003, 44, 1491–1498 CrossRef CAS.
- S. Kilic, S. Michalik, Y. Wang, J. K. Johnson, R. M. Enick and E. J. Beckman, Macromolecules, 2007, 40, 1332–1341 CrossRef CAS.
- B. Tan, H. M. Woods, P. Licence, S. M. Howdle and A. I. Cooper, Macromolecules, 2005, 38, 1691–1698 CrossRef CAS.
- Y. Wang, L. Hong, D. Tapriyal, I. C. Kim, I.-H. Paik, J. M. Crosthwaite, A. D. Hamilton, M. C. Thies, E. J. Beckman, R. M. Enick and J. K. Johnson, J. Phys. Chem. B, 2009, 113, 14971–14980 CrossRef CAS.
- M. R. Nelson and R. F. Borkman, J. Phys. Chem. A, 1998, 102, 7860–7863 CrossRef CAS.
- S. G. Kazarian, M. F. Vincent, F. V. Bright, C. L. Liotta and C. A. Eckert, J. Am. Chem. Soc., 1996, 118, 1729–1736 CrossRef CAS.
- H. Lee, E. Terry, M. Zong, N. Arrowsmith, S. Perrier, K. J. Thurecht and S. M. Howdle, J. Am. Chem. Soc., 2008, 130, 12242 CrossRef CAS.
- H. Chirowodza, M. X. Zou and R. D. Sanderson, J. Appl. Polym. Sci., 2010, 117, 3460–3465 CAS.
- C. E. Lipscomb and M. K. Mahanthappa, Macromolecules, 2009, 42, 4571–4579 CrossRef CAS.
- J. Eastoe, A. Paul, S. Nave, D. C. Steytler, B. H. Robinson, E. Rumsey, M. Thorpe and R. K. Heenan, J. Am. Chem. Soc., 2001, 123, 988–989 CrossRef CAS.
- C. F. Kirby and M. A. McHugh, Chem. Rev., 1999, 99, 565–602 CrossRef CAS.
- M. Destarac, C. Brochon, J. M. Catala, A. Wilczewska and S. Z. Zard, Macromol. Chem. Phys., 2002, 203, 2281–2289 CrossRef CAS.
- P. Licence, M. P. Dellar, R. G. M. Wilson, P. A. Fields, D. Litchfield, H. M. Woods, M. Poliakoff and S. M. Howdle, Rev. Sci. Instrum., 2004, 75, 3233–3236 CrossRef CAS.
- G. Moad and D. H. Solomon, Chemistry of free radical polymerization, Pergamon, 1995 Search PubMed.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. P. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160–1168 CrossRef CAS.
- M. F. Kemmere and T. Meyer, Supercritical Carbon Dioxide In Polymer Reaction Engineering, Wiley-VCH, 2006 Search PubMed.
- T. Sarbu, T. J. Styranec and E. J. Beckman, Ind. Eng. Chem. Res., 2000, 39, 4678–4683 CrossRef CAS.
- T. Berger, B. McGhee, U. Scherf and W. Steffen, Macromolecules, 2000, 33, 3505–3507 CrossRef CAS.
- M. L. O'Neill, M. Z. Yates, K. P. Johnston, C. D. Smith and S. P. Wilkinson, Macromolecules, 1998, 31, 2848–2856 CrossRef CAS.
- T. Carson, J. Lizotte and J. M. Desimone, Macromolecules, 2000, 33, 1917–1920 CrossRef CAS.
- P. Christian, S. M. Howdle and D. J. Irvine, Macromolecules, 2000, 33, 237–239 CrossRef CAS.
- Y. L. Hsiao, E. E. Maury, J. M. Desimone, S. Mawson and K. P. Johnston, Macromolecules, 1995, 28, 8159–8166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular weight distributions of homopolymers and copolymers. Proton NMR of typical copolymer stabiliser. Phase behaviour photographs. See DOI: 10.1039/c1py00062d |
| This journal is © The Royal Society of Chemistry 2011 |