DOI:
10.1039/C1PY00058F
(Review Article)
Polym. Chem., 2011,
2, 1597-1610
Recent advances in the chemistry of Group 4 metal complexes incorporating [OSSO]-type bis(phenolato) ligands as post-metallocene catalysts
Received
2nd February 2011
, Accepted 24th February 2011
First published on 16th March 2011
Abstract
Several recent efforts concerning post-metallocene Group 4 metal complexes incorporating mixed donor [OSSO]-type tetradentate bis(phenolato) ligands are reviewed. These [OSSO]-type ligands can be mainly classified as 5–5–5, 5–6–5 and 6–5–6 arrays of chelate rings toward central metals. As the main topics are described the design and unique coordination chemistry of the [OSSO]-type ligands and catalytic activities of the resultant titanium, zirconium, and hafnium complexes as single-site catalysts for polymerizations and copolymerizations of olefins.
 Norio Nakata | Norio Nakata, born in 1975, received his PhD from Kyoto University in 2003 under the supervision of Professor Norihiro Tokitoh. He became a postdoctoral fellow in 2003 and a research associate in 2004–2006 at the University of Tsukuba (Professor Akira Sekiguchi's group). In 2006, he joined Professor Akihiko Ishii's group as an assistant professor at Saitama University. His main research field is organometallic chemistry, and his current research interests are focused on the development of new post-metallocene catalysts. He received the ADEKA Co., Ltd., and Shionogi Co., Ltd., awards in Synthetic Organic Chemistry of Japan in 2008 and 2010, respectively. |
 Tomoyuki Toda | Tomoyuki Toda obtained his Masters degree of Science from the Graduate School of Science and Engineering, Saitama University, in 2010. During his Masters degree he studied the synthesis of Group 4 metal complexes with [OSSO]-type bis(phenolato) ligand and olefin polymerization catalyzed by the complexes. He has continued his research as a JSPS fellowship for young scientists since 2010. He is currently studying for a PhD under the supervision of Professor Akihiko Ishii and Assistant Professor Norio Nakata in the field of the synthesis and application of novel early transition metal complexes. |
 Akihiko Ishii | Akihiko Ishii received his PhD degree in 1987 from the University of Tokyo under the direction of Professor Naoki Inamoto. He was appointed as an Assistant Professor of the Department of Chemistry of Saitama University in 1987, and promoted to Associate Professor in 1994 and Professor in 2004. He was a visiting Professor of the University of Caen, France, 1997. He received the 1996 Progress Award of the Society of Synthetic Organic Chemistry, Japan. His research interest is in the area of synthesis and reactivities of organosulfur, organoselenium, and organometallic compounds. |
Introduction
Development of effective catalysts for olefin polymerization with controlled stereochemistry and narrow molecular weight distribution has been of great interest in both fundamental chemistry and industrial applications since the invention of homogeneous metallocene catalysts by Kaminsky and Sinn in 1980.1,2 In the last two decades, the so-called post-metallocene, early transition metal catalysts showing remarkable performances in activity and stereocontrol for olefin polymerization emerged.3–6 One of the important families of post-metallocene catalysts is that based on bis(phenolato) ligands bearing additional coordinating heteroatom moieties (Fig. 1). As a successful post-metallocene catalyst, Fujita developed Group 4 metal complexes supported by two monoanionic bidentate ligands such as the bis(phenoxy-imine) ligand (I).7–9 In the presence of methylaluminoxane (MAO) as the activator, these catalytic systems achieved ultrahigh activity for ethylene polymerization and syndiotactic living polymerization of propylene. Although these systems also showed high activity for polymerization of 1-hexene and other high α-olefins to yield high molecular weight poly(α-olefin)s, they were stereoirregular and regioirregular.
 |
| | Fig. 1 Examples of post-metallocene Group 4 complexes with chelating phenolato ligands. | |
To develop both active and stereoselective olefin polymerization catalysts, C2-symmetric bis(phenolato) ligand frameworks were designed, and several types of [ONNO]-type diamine bis(phenolato) tetradentate ligands have been investigated recently. Since a zirconium complex (II) featuring a linear [ONNO]-type Salan ligand that promotes the living polymerization of 1-hexene with high isotacticity at room temperature has been reported by Kolet al. in 2000,10 several related studies on Group 4 metal complexes bearing similar [ONNO]-type bis(phenolato) ligands have been published to date. However, these [ONNO]-type catalysts showed only poor activity in α-olefin isoselective polymerization because of too strong coordination of the nitrogen atoms to the metal center.11–19 In contrast to strong hard-donor nitrogen atoms in the [ONNO]-type ligands, sulfur is a soft-donor and is expected to interact more weakly with hard early transition metal cationic centers to increase the activity for α-olefin polymerization. Thus, complexes based on a sulfur-linked bis(phenolato) [OSSO]-type ligand have been introduced into olefin polymerization. This effect of sulfur has been proposed to account experimentally20–22 and theoretically23,24 for the highly active ethylene polymerization catalyzed by some titanium and zirconium complexes having sulfur-bridged [OSO]-type tridentate bis(phenolato) ligands (III) in comparison with the corresponding methylene-bridged or biaryloxide analogues. As a pioneering study on [OSSO]-type ligands, Group 4 metal complexes with 1,4-dithiabutanediyl-linked [OSSO]-type bis(phenolato) ligand featuring a 5–5–5 array of chelate rings were reported by Okuda et al. in 2003 and were able to efficiently polymerize styrene upon activation with MAO to give isotactic polystyrene.25 In the present review, we outline the preparations and unique coordination chemistry of a new family of ligands, [OSSO]-type tetradentate dianionic ligands. We also describe the catalytic ability of Group 4 metal complexes incorporating these [OSSO]-type bis(phenolates) as ancillary ligands for polymerizations of olefins.
Design and synthesis of the [OSSO]-type bis(phenolato) ligands
[OSSO]-Type bis(phenolato) ligands reported are structurally classified as three families depending on chelating ring sizes toward the metal center. The first family is based on a compact 5–5–5 array of chelate rings. Okuda et al. prepared a series of 1,4-dithiabutanediyl-linked [OSSO]-type bis(4,6-disubstituted phenolato) ligands (1a: R1, R2 = H, 1b: R1 = H, R2 = Me, 1c: R1, R2 = Me, 1d: R1 = iPr, R2 = tBu, 1e: R1 = tBu, R2 = Me, 1f: R1, R2 = tBu, 1g: R1 = tBu, R2 = OMe, 1h: R1 = CMe2Ph, R2 = Me, and 1i: R1, R2 = CMe2Ph) by the nucleophilic substitution of 1,2-dibromoethane with 3,5-disubstituted 2-hydroxybenzenethiol in the presence of NaOH (Scheme 1).25,26 They also succeeded in the synthesis of trans-1,2-dithiocyclohexanediyl-bridged [OSSO]-type ligands with 5–5–5 chelation rings (2a: R1 = H, R2 = Me, 2b: R1, R2 = iPr, 2c: R1 = tBu, R2 = Me, and 2d: R1, R2 = tBu) as a racemic form in three steps (Scheme 2).27,28 The synthesis of 2 features the ring opening of cyclohexene oxide by the arenethiolate, followed by chlorination of the resulting alcohol with thionyl chloride in dichloromethane to give the corresponding (trans-2-chlorocyclohexylthio)phenol derivative. Finally, the substitution by a second equivalent of the thiolate affords the racemic 2, where the exclusive formation of the trans-form was explained in terms of the anchimeric effect of the thioarene moiety. Chiral resolution of rac-2d (R1, R2 = tBu) was achieved through separation of the derived bis[(1S)-camphorsulfonates] diastereomers by fractional crystallization and column chromatography followed by hydrolysis. The absolute configuration of each diastereomer was determined by X-ray crystallography.27 In addition, enantiopure (S,S)-2c and (R,R)-2c can be obtained by preparative chiral HPLC.28
![Synthesis of 5–5–5 [OSSO]-type ligands.](/image/article/2011/PY/c1py00058f/c1py00058f-s1.gif) |
| | Scheme 1 Synthesis of 5–5–5 [OSSO]-type ligands. | |
![Synthesis of racemic trans-1,2-dithiocyclohexanediyl-bridged 5–5–5 [OSSO]-type ligands.](/image/article/2011/PY/c1py00058f/c1py00058f-s2.gif) |
| | Scheme 2 Synthesis of racemic trans-1,2-dithiocyclohexanediyl-bridged 5–5–5 [OSSO]-type ligands. | |
The second family is based on [OSSO]-type ligands featuring 5–6–5 array of chelate rings. Similar to the synthetic method of the 5–5–5 [OSSO]-type ligand 1, 1,5-dithiapentanediyl-linked bis(4,6-disubstituted phenolato) ligands (3a: R1 = tBu, R2 = Me, 3b: R1, R2 = tBu, 3c: R1 = tBu, R2 = OMe) are prepared by treatment of 1,3-dibromopropane with 3,5-disubstituted 2-hydroxybenzenethiol in the presence of NaOH (Scheme 3).25,26
![Synthesis of 5–6–5 [OSSO]-type ligands.](/image/article/2011/PY/c1py00058f/c1py00058f-s3.gif) |
| | Scheme 3 Synthesis of 5–6–5 [OSSO]-type ligands. | |
On the other hand, the third family is based on [OSSO]-type ligands with 6–5–6 chelate rings, which are sulfur-analogues of the [ONNO]-type Salan ligands (see the ligand of II in Fig. 1). Kolet al. reported the development of a 1,4-dithiabutanediyl-linked [OSSO]-type bis(phenolato) ligand with 6–5–6 chelate rings (4), which was synthesized by the reaction of ethanedithiol with 2 equiv. of 2-(bromomethyl)-4,6-di-tert-butylphenol on a multigram scale (Scheme 4).29 In 2009, we also demonstrated a 6–5–6 [OSSO]-type ligand featuring the fusion of a trans-cyclooctane-1,2-diyl ring (5) as a racemic form (Scheme 5).30 According to a modified Kol's method, ligand 5 was readily prepared by the reaction of trans-cyclooctane-1,2-dithiol with 2 equiv. of 2-(bromomethyl)-4,6-di-tert-butylphenol in the presence of Et3N.
![Synthesis of 6–5–6 [OSSO]-type ligand 4.](/image/article/2011/PY/c1py00058f/c1py00058f-s4.gif) |
| | Scheme 4 Synthesis of 6–5–6 [OSSO]-type ligand 4. | |
![Synthesis of 6–5–6 [OSSO]-type ligand 5 bearing a trans-cyclooctane-1,2-diyl ring.](/image/article/2011/PY/c1py00058f/c1py00058f-s5.gif) |
| | Scheme 5 Synthesis of 6–5–6 [OSSO]-type ligand 5 bearing a trans-cyclooctane-1,2-diyl ring. | |
Group 4 metal complexes incorporating [OSSO]-type ligands: synthesis and structures
The synthesis of Group 4 metal complexes bearing [OSSO]-type ligands was accomplished by treating [OSSO]-type bis(phenol)s 1–5 with suitable metal precursors in benzene or toluene. A series of titanium dichloro and di(isopropoxy) complexes 6 and 7 with 5–5–5 [OSSO]-type ligands 1 were synthesized by treating ligands 1 with the titanium precursor TiL4 (L = Cl, OiPr) in toluene (Scheme 6).25,26,31 The NMR spectra of dichloro complexes 6a–d with small ortho substituents (R1 = H, Me, iPr) are in agreement with a C2-symmetric, helical structure, but the complexes become fluxional at higher temperatures. The NMR spectra of the corresponding di(isopropoxy) complexes 7a–d are fluxional in solution even at room temperature due to rapid interconversion between the Δ and Λ isomers, possibly via a tetrahedrally coordinated transition state 7′ (Scheme 7).26,31 In contrast, both dichloro and di(isopropoxy) complexes 6e–i and 7e–i with bulky ortho-substituents (R1 = tBu, CMe2Ph) exhibit a rigid C2-symmetric, helical structure in solution up to 100 °C. In the X-ray crystallographic analyses for two dichloro complexes 6e and 6i, the titanium core lay at the centre of a distorted octahedral coordination sphere located cis to sulfur and trans to oxygen dispositions, adopting a cis-α configuration. In the case of 1,2-dithiocyclohexanediyl-bridged 5–5–5 [OSSO]-type ligand 2, complexation of racemic 2c and 2d with TiCl4 in pentane proceeded in a diastereoselective fashion to give only the corresponding titanium chloro complexes with the (Λ*,R*,R*)-configuration (8c: R1 = tBu, R2 = Me, 8d: R1, R2 = tBu) (the (Δ*,R*,R*)-forms were not obtained) (Scheme 8).27,28
![Synthesis of titanium dichloro (6) and di(isopropoxy) (7) complexes with 5–5–5 [OSSO]-type ligands 1.](/image/article/2011/PY/c1py00058f/c1py00058f-s6.gif) |
| | Scheme 6 Synthesis of titanium dichloro (6) and di(isopropoxy) (7) complexes with 5–5–5 [OSSO]-type ligands 1. | |
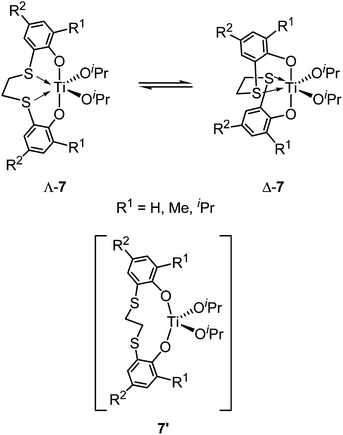 |
| | Scheme 7 Interconversion between Λ-7 and Δ-7. | |
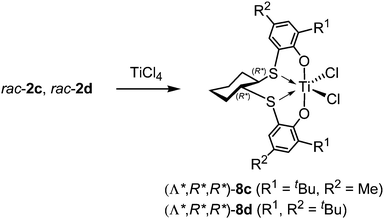 |
| | Scheme 8 Diastereoselective formation of (Λ*,R*,R*)-8c and (Λ*,R*,R*)-8d by the reaction of rac-2c and rac-2d, respectively, with TiCl4. | |
Titanium dibenzyl complex 9 having a 5–5–5 [OSSO]-type ligand was prepared by treating the corresponding titanium dichloro complex 6 with an ethereal solution of benzylmagnesium chloride in toluene at −30 °C (Scheme 9).31,32 Although complex 9b, which does not have bulky ortho-substituents, was found to be fluxional and unstable in solution, variable temperature NMR studies on complexes 9e and 9f (−70 to +90 °C) indicated that both complexes are thermally robust and maintain their chiral C2-symmetric structures within this temperature range. The molecular structure of 9f in the crystalline state was determined by X-ray analysis to have a C2-symmetric [OSSO]-type ligand with two benzyl ligands in the cis-position. One of the Ti–Cbenzyl–Cipso bond angles in 9f is characteristically narrower than the other, suggesting η2-coordination of the benzyl ligand in the crystalline state.
![Synthesis of titanium dibenzyl complexes 9 bearing 5–5–5 [OSSO]-type ligands.](/image/article/2011/PY/c1py00058f/c1py00058f-s9.gif) |
| | Scheme 9 Synthesis of titanium dibenzyl complexes 9 bearing 5–5–5 [OSSO]-type ligands. | |
Zirconium (10) and hafnium (11) dibenzyl complexes incorporating 5–5–5 [OSSO]-type ligand 1e are also synthesized by the reaction of 1e with M(CH2Ph)4 (M = Zr, Hf) in quantitative yields (Scheme 10).25 Complexes 10 and 11 have molecular C2-symmetry, which was evident from the symmetry-related phenolate rings and the presence of only two AB patterns due to CH2 units of the bridged methylenes and two cis-benzyl ligands in the NMR study. Crystallographic structure determination of the hafnium complex 11 confirmed that this complex took a C2-symmetric cis-α configuration with two benzyl ligands and thioether groups.
![Synthesis of zirconium (10) and hafnium (11) dibenzyl complexes by the reaction of 5–5–5 [OSSO]-type ligands 1e with M(CH2Ph)4 (M = Zr, Hf).](/image/article/2011/PY/c1py00058f/c1py00058f-s10.gif) |
| | Scheme 10 Synthesis of zirconium (10) and hafnium (11) dibenzyl complexes by the reaction of 5–5–5 [OSSO]-type ligands 1e with M(CH2Ph)4 (M = Zr, Hf). | |
Reactions of 5–6–5 [OSSO]-type ligands 3a–c with TiL4 (L = Cl, OiPr) in toluene afforded the corresponding dichloro and di(isopropoxy) complexes (12a: R1 = tBu, R2 = Me, 12b: R1, R2 = tBu, 12c: R1 = tBu, R2 = OMe, 13a: R1 = tBu, R2 = Me, 13b: R1, R2 = tBu, 13c: R1 = tBu, R2 = OMe) in quantitative yields (Scheme 11).26,31 In NMR study, while dichloro complexes 12a–c showed a pattern of broad signals at ambient temperature owing to the fluxional behavior in the NMR time scale, the low-temperature 1H NMR spectra exhibited the six CH2 protons of the backbone as six broad signals, indicating an unsymmetrical structure. In contrast, titanium di(isopropoxy) complexes 13a–c are configurationally fluxional in the temperature range of −80 to +80 °C, exhibiting apparent Cs-symmetry in solution. A crystallographic analysis revealed that 12b adopted a C1-symmetric structure in which one sulfur atom was disposed trans to a chloro ligand and another sulfur atom trans to an oxygen atom, resulting in cis-coordinated phenolato groups. The structure of 13b, which was revealed to take a cis-α configuration (trans-O,O, cis-S,S) in the crystalline state, is distinct from that of the dichloro complex 12b, indicating that more than one configuration is accessible for the complexes bearing 5–6–5 [OSSO]-type ligand 3b.
![Synthesis of titanium dichloro (12) and di(isopropoxy) (13) complexes with 5–6–5 [OSSO]-type ligands from 3.](/image/article/2011/PY/c1py00058f/c1py00058f-s11.gif) |
| | Scheme 11 Synthesis of titanium dichloro (12) and di(isopropoxy) (13) complexes with 5–6–5 [OSSO]-type ligands from 3. | |
[OSSO]-Type ligand precursor 4 having 6–5–6 chelate rings reacted with titanium and zirconium alkoxides (Ti(OiPr)4 and Zr(OtBu)4) in diethyl ether to give the corresponding dialkoxy complexes 14 and 15 in practically quantitative yields (Scheme 12).29Titanium di(isopropoxy) complex 15 appears to be C2v-symmetric at room temperature in the observation of sharp singlets of the methylene groups in the 1H NMR spectrum, whereas zirconium di(tert-butoxy) complex 14 shows a C2-symmetric [OSSO]-type ligand in the 1H NMR spectrum at room temperature, as is evident from the AB pattern of S-benzyl protons. Similar to the case of Okuda's zirconium dibenzyl complex 10, 6–5–6 [OSSO]-type ligand 4 was treated with Zr(CH2Ph)4 in toluene to give the corresponding zirconium dibenzyl complex 16 (Scheme 12).29
![Synthesis of titanium (14 and 15) and zirconium (16) complexes with 6–5–6 [OSSO]-type ligand 4.](/image/article/2011/PY/c1py00058f/c1py00058f-s12.gif) |
| | Scheme 12 Synthesis of titanium (14 and 15) and zirconium (16) complexes with 6–5–6 [OSSO]-type ligand 4. | |
In the case of the 6–5–6 [OSSO]-type ligand 5 based on a trans-1,2-cyclooctanediyl platform, the same type of zirconium dibenzyl complex 17 was prepared by the reaction of 5 with Zr(CH2Ph)4 in toluene (Scheme 13).30Zirconium dichloro complex 18 was also synthesized by deprotonation of ligand 5 with 2 equiv. of BuLi in diethyl ether at 0 °C followed by treatment with ZrCl4 at −78 °C (Scheme 14).33NMR spectroscopic data of 17 and 18 exhibited the equivalency of the two phenolate ligands as well as the two benzyl moieties in 17, indicating that 17 and 18 appear to be C2-symmetric in solution in the NMR time scale as similar as above-mentioned complexes bearing [OSSO]-type ligands. The molecular structure analyses of 17 and 18 by X-ray revealed that each zirconium core lay at the centre of a distorted octahedral coordination sphere with two sulfur and two oxygen atoms at the cis and trans dispositions, respectively, adopting a cis-α, (Λ*,S*,S*)-configuration.
![Synthesis of zirconium dibenzyl complex 17 by the reaction of with 6–5–6 [OSSO]-type ligand 5 with Zr(CH2Ph)4.](/image/article/2011/PY/c1py00058f/c1py00058f-s13.gif) |
| | Scheme 13 Synthesis of zirconium dibenzyl complex 17 by the reaction of with 6–5–6 [OSSO]-type ligand 5 with Zr(CH2Ph)4. | |
![Synthesis of zirconium dichloro complex 18 bearing 6–5–6 [OSSO]-type ligand from 5.](/image/article/2011/PY/c1py00058f/c1py00058f-s14.gif) |
| | Scheme 14 Synthesis of zirconium dichloro complex 18 bearing 6–5–6 [OSSO]-type ligand from 5. | |
Catalytic activity in polymerizations
As mentioned above, almost Group 4 metal complexes incorporating [OSSO]-type bis(phenolato) ligands often take an octahedral configuration, providing opportunities to inspect whether or not the symmetry rule for the tetrahedral-type metallocene complex2,3 still holds true for these configurationally and electronically very different complexes. Okuda and Proto discovered that titanium complexes 6f and 7f with bulky tBu substituents at ortho positions in the aromatic rings of the 5–5–5 [OSSO]-type ligand polymerized styrene into isotactic polymers (6f: Mn = 265.4 × 104 g mol−1, Mw/Mn = 2.0, and Tm = 223 °C, 7f: Mn = 171.8 × 104 g mol−1, Mw/Mn = 1.8, and Tm = 223 °C) with high activities (6f: 1543 g per mmol (6f) per h, 7f: 571 g per mmol (7f) per h) upon activation with MAO at 40 °C in toluene (1 × 10−4 mol of complex, Al/Ti = 1500) (Scheme 15).25,26,31 In addition, Proto reported the isospecific polymerization of several para-substituted styrenes catalyzed by 6f/MAO (1 × 10−6 mol of 6f, Al/Ti = 500) in toluene at 25 °C.34 The relative rates for polymerizations appeared to decrease in the following order: p-methylstyrene (4.15) > styrene (1) > p-tert-butylstyrene (0.60) > p-bromostyrene (0.021) > p-chlorostyrene (0.019). These results show that the interaction of the aromatic ring with the metal is involved in the polymerization mechanism and plays a crucial role in determining the catalyst activity. The complexes of zirconium and hafnium, 10 and 11, bearing the same 5–5–5 [OSSO]-type ligand also polymerized styrene isospecifically (10: Mn = 16.3 × 104 g mol−1, Mw/Mn = 1.9, and Tm = 218 °C, 11: Mn = 4.0 × 104 g mol−1, Mw/Mn = 1.9, and Tm = 220 °C), albeit with low activity (10: 22 g per mmol (10) per h, 11: 5 g per mmol (11) per h).25,31 However, the catalyst precursors with less bulky ortho-substituents such as H, Me, and iPr groups 6a–d produced atactic polystyrene with low activities and with broad molecular weight distributions upon activation with MAO.26 On the other hand, the titanium complexes 12a and 13c having 1,5-dithiapentanediyl-linked 5–6–5 [OSSO]-type ligand 3 activated by MAO at 40 °C in toluene produced syndiotactic polystyrenes with quite low activity (12a: 4 g per mmol (12a) per h, 13c: 5 g per mmol (13c) per h) (Scheme 15).26 The resulting syndiotactic polystyrenes show broad or bimodal molecular weight distributions (12a: Mn = 0.7 × 104 g mol−1, Mw/Mn = 2.89, 13c: Mn = 89.1 × 104 and 0.5 × 104 g mol−1, Mw/Mn = 1.78 and 1.71) and somewhat higher melting temperature Tm of 268 °C (12a) compared with those of isotactic polystyrene (ca. 220 °C).
 |
| | Scheme 15 Isotactic or syndiotactic polymerization of styrene catalyzed by titanium complexes 6f, 7f, 12a, and 13c activated by MAO. | |
The titanium dichloro complex 6f activated by MAO (1 × 10−5 mol of 6f, Al/Ti = 500) can also be a catalyst for ethylene polymerization at temperatures between 25 and 80 °C in toluene (ethylene 2–6 bar).35 This catalyst system based on 6f shows a relatively high activity up to 6200 g per mmol (6f) per h per atm. The thermograms of the obtained polymers indicated two transitions (Tm = 118–121 and 129–132 °C), one of which is significantly lower than the values for linear polyethylenes. As evidenced by 13C NMR spectroscopic data, the polymers have ethyl, butyl, and longer-chain branches in a range variable from 0.2 to 2.0%. The GPC analyses of the polymers showed broad molecular weight distributions (Mw/Mn = 13–65) except the polymer obtained by using a tenth amount of the 6f/MAO catalyst system (1 × 10−6 mol of 6f, Al/Ti = 5000). The 6f/MAO catalyst system is also active in the trans-1,4 selective polymerization of conjugated dienes such as butadiene (87.2–95.6%) and isoprene (80–91%).36 The molecular weight distributions of the polymers are monomodal with the polydispersity indexes (butadiene: Mn = 2700–5700 g mol−1, Mw/Mn = 2.0–2.9, isoprene: Mn = 4600–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1, Mw/Mn = 1.4–2.3), consistent with a single site behavior of the catalyst.
100 g mol−1, Mw/Mn = 1.4–2.3), consistent with a single site behavior of the catalyst.
The titanium dibenzyl complex 9e activated with [PhNMe2H][B(C6F5)4] in the presence of the trioctylaluminium compound AlOct3 achieved for the first time the living polymerization of styrene with perfect isotacticity at 25 °C (mm > 95%, judging from the 13C NMR spectra) (1 × 10−3 M of 9e, 0.9 × 10−3 M of activator in 10 mL of toluene).32 The resulting isotactic polystyrenes showed narrow molecular weight distributions (Mn = 67000–106100 g mol−1, Mw/Mn < ∼1.2) and slightly lower melting temperature Tm of ca. 210 °C than those of higher molecular-weight isotactic polystyrene.
The racemic and optically active precatalyst 8d having a 1,2-dithiocyclohexanediyl-bridged 5–5–5 [OSSO]-type ligand 2d is also capable of polymerizing styrene with isoselectivity (Scheme 16).27,28 When treated with MAO in toluene at 40 °C (1.25 × 10−6 mol of 8d, Al/Ti = 1500), the racemic and optically active complex 8d polymerized styrene with activities up to 146 g per mmol (8d) per h, which are somewhat lower than those of precatalysts 6f and 7f. Furthermore, Okuda has been succeeded in the first homochiral isotactic polymerization of styrene with enantiopure precatalysts (+)-(Δ,S,S)-8c and (+)-(Δ,S,S)-8d by utilizing chain-transfer methodology in the presence of 1-hexene.27,28 For the complex (+)-(Δ,S,S)-8d it was established that by variation of the 1-hexene/styrene ratio the Mn values of the oligostyrenes could be controlled down to ca. 750 g mol−1. In a similar set of oligomerization experiments, (+)-(Δ,S,S)-8c produced significantly higher molecular weight oligomers (Mn > 1290 g mol−1). A typical pair of obtained oligomers having Mn values of approximately 2000 g mol−1 showed peaks assignable to chains containing one to five 1-hexene units (m = 0–4) attached to a polystyrene segment (maximum at n ≈ 30) in the MALDI TOF mass spectra. NMR analyses confirmed the presence of isotactic polystyrene segments terminated by regio- and stereoirregular oligo(hexene) segments. The main-chain chirality of produced oligostyrene results in optical activity up to a degree of polymerization of 45 for styrene by dependence of the specific rotation [α]D23 and molar rotation [Φ]D23 on Mn values of obtained oligostyrene terminated by 1-hexene. Thus, beyond Mn ≈ 5000 g mol−1 optical activity cannot be reliably measured, rendering higher molecular weight polystyrenes cryptochiral.
The isotactic polymerization of 4-methyl-1,3-pentadiene (4-MPD) to produce 1,2-polymer has been performed with various heterogeneous titanium and vanadium catalysts activated by AlEt3.37 As an example of homogeneous catalyst systems, the titanium dichloro complex 6f was first used to promote the isospecific polymerization of 4-MPD with MAO as the activator (1 × 10−5 mol of 6f, Al/Ti = 2200) (Scheme 17).38 The 13C NMR spectrum of the obtained polymer displayed highly stereoregular polymer where the monomers were connected predominantly in the 1,2-fashion. The small signals due to the corresponding 4-MPD units in 1,4-arrangement were estimated from 13C NMR by amount less than 3%. X-Ray diffraction pattern and IR spectrum of this polymer were close to those reported for 1,2-poly(4-MPD) obtained in the presence of heterogeneous titanium and vanadium catalysts.37 The GPC analysis of the resulting 1,2-poly(4-MPD) showed Mw = 57000 and a narrow polydispersity (Mw/Mn) of 1.7. In the DSC measurement, the lower melting temperature Tm of 146 °C was attributed to the incorporation of some 1,4-units, which disturb the crystallinity of the polymer even in small amounts.
 |
| | Scheme 17 Isotactic polymerization 4-methyl-1,3-pentadiene catalyzed by the 6f/MAO system. | |
The titanium precatalyst 6f can be expanded to olefin copolymerization (Scheme 18). Thus, the copolymerization of styrene with a small amount of ethylene (1 atm) employing the system consisting of complex 6f/MAO in toluene (1 × 10−6 mol of 6f, Al/Ti = 900) resulted in the formation of isotactic polystyrene containing a low amount of isolated ethylene units.39,40 The 13C NMR spectroscopic study on this ethylene–styrene copolymer showed that an “enantiomorphic site” control mechanism is operating for isotactic propagation to produce mmMmm arrangement copolymer. DSC measurement also indicated the presence of isolated ethylene units modifying the crystallization behavior of the isotactic polystyrene. The styrene polymerization in the presence of propylene (1 bar) using 6f/MAO (2.5 × 10−5 mol of 6f, Al/Ti = 500) also produced iPS-block-iPP-block-iPS copolymers (Tm = 171/192 °C) containing long isotactic styrene sequences interrupted by short isotactic propylene strings.41 The microstructure of this copolymer elucidated by 13C NMR analysis showed that the opposite regiochemistry of insertion of the monomers was retained in the copolymerization, producing tail-to-tail and head-to-head linkages between the homopolymer blocks. The GPC analysis of the polymer showed a monomodal molecular weight distribution (Mw = 33000 g mol−1) and a narrow polydispersity (Mw/Mn) of 1.9. In addition, Proto demonstrated the alternating ethylene–butadiene copolymerization promoted by 6f/MAO in toluene at 0–50 °C (1 × 10−6 mol of 6f, Al/Ti = 1000).42 This catalyst system exhibited good activity (96–1332 g per mmol (6f) per h), which is more than 4 times higher than those reported in the case of neodymocene systems. The alternating microstructural features of resulting copolymers were confirmed by 13C NMR spectroscopy. Butadiene units contained in the range from 9% to 65% in the polymers are mainly inserted in the 1,4-trans configuration with a minor amount of 1,4-cis units. Notably neither 1,2-units nor the formation of cycle by butadiene cyclopolymerization was detected. The molecular weight distributions are monomodal ranging between 1.9 and 3.4 (Mw = 16000–64000 g mol−1), indicating a single-site catalyst. Moreover, the copolymerization of ethylene with 4-MPD catalyzed by 6f/MAO or 6h/MAO in toluene (1 × 10−5 mol of 6f or 6h, Al/Ti = 1200) produced exclusively ethylene–(4-MPD) copolymers with good activity (6f: 128–298 g per mmol (6f) per h, 6h: 60–411 g per mmol (6h) per h).43 The catalyst 6h incorporated more efficiently the 4-MPD in the polymer, resulting in the corresponding copolymers with a high content of 4-MPD up to 83%. The Tg values increased by increasing the 4-MPD content varying from −37.9 to 2.4 °C while the Tm values increased by increasing the ethylene content in the copolymer from 85.3 to 115.6 °C. The molecular weight of the polymers did not vary significantly in the range of composition explored for 6f/MAO system (Mn = 4900–7100 g mol−1), whereas increase of one order of magnitude for 6h/MAO system was observed with increasing the 4-MPD content from 40% (Mn = 10400 g mol−1) to 83% (Mn = 104000 g mol−1). The molecular weight distributions were monomodal for both systems ranging between 1.31 and 2.11. Quite recently, Proto also reported on the ability of these catalyst systems (1 × 10−5 mol of 6f or 6h, Al/Ti = 1000, in toluene, at −30 to 70 °C) to promote the copolymerization of ethylene with isoprene producing copolymers with predominantly trans-1,4 microstructure and isoprene content ∼60%.44 The produced copolymers are white elastomers and the Tg values are varied by increasing the isoprene content ranging from −87.0 to −70 °C.
Proto and Capacchione et al. succeeded in the living stereoregular polymerization of styrene and butadiene promoted by the titanium complex 6h with bulky cumyl groups at ortho positions in the aromatic rings activated by MAO (Scheme 19).45 The homopolymerizations of styrene and butadiene catalyzed by 6h/MAO system under optimized conditions (styrene: 1 × 10−6 mol of 6h, Al/Ti = 1200, in toluene, butadiene: 1 × 10−5 mol of 6h, Al/Ti = 1200, in toluene) afforded highly isotactic polystyrene (iPS) and high 1,4-trans-polybutadiene (PB), respectively. The narrow molecular weight distribution and the observation that Mn increases linearly with styrene conversion clearly indicate a living behavior of polymerization at room temperature (Mw/Mn = 1.05–1.19). A similar behavior was also observed in the case of butadiene polymerization at 0 °C producing 1,4-trans-PB with a minor amount of cis-1,4-units (<5%) but in this case a slight broadening of the molecular weight distribution with conversion was confirmed (Mw/Mn = 1.28–1.77). The copolymerization of styrenevia sequential addition of butadiene using 6h/MAO in toluene at 25 °C (1 × 10−6 mol of 6h, Al/Ti = 1200) resulted in the formation of iPS-block-1,4-trans-PB copolymer. The obtained block copolymers exhibited high molecular weight (PS: Mn = 108000–171000 g mol−1, PS-b-PB: Mn = 139000–201000 g mol−1) and narrow molecular weight distribution, confirming for both the initial iPS homopolymer unit (Mw/Mn = 1.04–1.08) and the final iPS-block-1,4-trans-PB copolymer unit (Mw/Mn = 1.07–1.11) a living behavior. The comparison of DSC scans showed that both blocks in copolymer are crystalline with Tm values (PS: 222.8–224.1 °C, PB: 40.4/82.9–42.6/85.9 °C) similar to that of iPS (224.5 °C) and 1,4-trans-PB (46.9/87.8 °C).
Okuda also reported that Group 4 metal complexes featuring 5–5–5 [OSSO]-type ligand 1f efficiently catalyzed 1-hexene oligomarization and that the regioselectivity of insertion was switched upon changing the metal from titanium to zirconium or hafnium (Scheme 20).46 Thus, oligomerization of 1-hexene using titanium dimethyl complex 19 or dibenzyl complex 9f activated by one equivalent of B(C6F5)3 proceeded at room temperature within 5 min to yield oligo(1-hexene)s (19: Mn = 352 g mol−1, Mw/Mn = 1.30, 9f: Mn = 352 g mol−1, Mw/Mn = 1.33) with turnover frequency (TOF) > 19960–20700 h−1. 1H NMR spectra of the resulting oligo(1-hexene)s indicated the presence of internal vinylene moieties E- and Z-RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR′, suggesting 2,1-regioselectivity (92–99%). In contrast, zirconium and hafnium dibenzyl complexes 10 and 11, respectively, were found to be less active by two and four orders of magnitude, respectively, than the corresponding titanium homologues 9f and 19 (10: Mn = 224 g mol−1, Mw/Mn = 1.23, TOF = 196 h−1, 11: Mn = 338 g mol−1, Mw/Mn = 1.38, TOF = 3.8 h−1). The 1H NMR spectra showed the selective formation of oligo(1-hexene)s with the terminal vinylidene moiety RR′C = CH2 (10: 95% and 11: 93%).
CHR′, suggesting 2,1-regioselectivity (92–99%). In contrast, zirconium and hafnium dibenzyl complexes 10 and 11, respectively, were found to be less active by two and four orders of magnitude, respectively, than the corresponding titanium homologues 9f and 19 (10: Mn = 224 g mol−1, Mw/Mn = 1.23, TOF = 196 h−1, 11: Mn = 338 g mol−1, Mw/Mn = 1.38, TOF = 3.8 h−1). The 1H NMR spectra showed the selective formation of oligo(1-hexene)s with the terminal vinylidene moiety RR′C = CH2 (10: 95% and 11: 93%).
 |
| | Scheme 20 Oligomerization of 1-hexene catalyzed by titanium complexes 9f and 19, zirconium complex 10, and hafnium complex 11 activated by B(C6F5)3. | |
In contrast to 1-hexene oligomarization using Okuda's titanium complexes 9f and 19 featuring 5–5–5 [OSSO]-type ligand 1f, zirconium complexes 16 and 17 incorporating 6–5–6 [OSSO]-type ligands 4 or 5 achieved the polymerization of 1-hexene (Scheme 21).29,30 Kol's complex 16 upon activation with B(C6F5)3 is an active 1-hexene polymerization catalyst at room temperature, leading to a stereoirregular (atactic) polymer despite its C2 symmetry.29 The GPC analysis of the resulting polymer revealed a relatively low molecular weight of 7400 g mol−1 and a relatively narrow polydispersity (Mw/Mn) of 1.6, indicating a single-site catalyst. This catalyst system showed an activity value of 80 g per mmol (16) per h. On the other hand, upon activation with B(C6F5)3 or (Ph3C)[B(C6F5)4], the polymerization of 1-hexene using 17 in benzene/hexane at room temperature or 0 °C proceeded vigorously and exothermically to give poly(1-hexene)s with larger molecular weights (Mw = 41000–120000 g mol−1) and narrow polydispersities (Mw/Mn) ranging between 1.6 to 2.1.30 Surprisingly, in spite of such exothermic reactions, the 13C NMR spectra of the poly(1-hexene)s showed excellent isotacticity (mmmm > 95%) in all cases. It is noteworthy that the activity of the 17/(Ph3C)[B(C6F5)4] system was evaluated to be 2500 g per mmol (17) per h, which is more than 30 times larger than those of single-site zirconium complexes having 6–5–6 [ONNO]-10–16 and 6–5–6 [OSSO]-type ligands.29
![Polymerization of 1-hexene catalyzed by zirconium dibenzyl complexes 16 and 17 activated by B(C6F5)3 or (Ph3C)[B(C6F5)4].](/image/article/2011/PY/c1py00058f/c1py00058f-s21.gif) |
| | Scheme 21
Polymerization of 1-hexene catalyzed by zirconium dibenzyl complexes 16 and 17 activated by B(C6F5)3 or (Ph3C)[B(C6F5)4]. | |
Table 1 summarizes the results of olefin polymerizations mentioned in this review.
|
Chelate rings |
No. |
ML2 |
Activator |
Olefin
|
M
n (or Mw*)/g mol−1 |
M
w/Mn |
T
m/°C |
Activitya |
Tacticityb |
Conditions; comments |
Ref. |
|
g per mmol (cat) per h.
Abbreviations: iso: isotactic; a: atactic; syndio: syndiotactic.
[PhNMe2H][B(C6F5)4].
4-Methyl-1,3-pentadiene.
(Ph3C)[B(C6F5)4].
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Styrene
|
265.4 × 104 |
2.0 |
223 |
1543 |
iso
|
40 °C in toluene, 1 × 10−4 mol of 6f, Al/Ti = 1500 |
25,26, and 31
|
| 5–5–5 |
7f
|
Ti(OiPr)2 |
MAO
|
Styrene
|
171.8 × 104 |
1.8 |
223 |
571 |
iso
|
40 °C in toluene, 1 × 10−4 mol of 7f, Al/Ti = 1500 |
25,26, and 31
|
| 5–5–5 |
10
|
ZrBn2 |
MAO
|
Styrene
|
16.3 × 104 |
1.9 |
218 |
22 |
iso
|
|
25 and 31
|
| 5–5–5 |
11
|
HfBn2 |
MAO
|
Styrene
|
4.0 × 104 |
1.9 |
220 |
5 |
iso
|
|
25 and 31
|
| 5–5–5 |
6a–d
|
TiCl2 |
MAO
|
Styrene
|
|
|
|
|
a
|
Low activity and broad molecular weight distribution |
26
|
| 5–6–5 |
12a
|
TiCl2 |
MAO
|
Styrene
|
0.7 × 104 |
2.89 |
268 |
4 |
syndio
|
40 °C in toluene |
26
|
| 5–6–5 |
13c
|
Ti(OiPr)2 |
MAO
|
Styrene
|
89.1 × 104 |
1.78 |
|
5 |
syndio
|
40 °C in toluene; bimodal |
26
|
| 0.5 × 104 |
1.71 |
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Ethylene
|
|
13–65 |
118–121 |
<6200 |
|
1 × 10−5 mol of 6f, Al/Ti = 500, 25–80 °C in toluene, ethylene (2–6 atm); branched polyethylene |
35
|
| 129–132 |
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Butadiene
|
2700–5700 |
2.0–2.9 |
|
|
|
trans-1,4 selective (87.2–95.6%) |
36
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Isoprene
|
4600–25100 |
1.4–2.3 |
|
|
|
trans-1,4 selective (80–91%) |
36
|
| 5–5–5 |
9e
|
TiBn2 |
Borate
c–AlOct3 |
Styrene
|
67000–106100 |
< ∼1.2 |
ca. 210 |
|
iso
|
1 × 10−3 mol of 9e, 0.9 × 10−3 M of the activator, 25 °C in toluene; living polymerization, mm > 95%, |
32
|
| 5–5–5 |
8d
|
TiCl2 |
MAO
|
Styrene
|
|
|
<146 |
|
|
1.25 × 10−6 mol of 8d, Al/Ti = 1500, 40 °C in toluene |
27 and 28
|
| 5–5–5 |
(+)-8d |
TiCl2 |
MAO
|
1-Hexene–styrene |
ca. 750 |
|
|
|
|
Oligomerization, isotactic PS segments terminated by region- and stereoirregular oligo(1-hexene) segments, optically active (Mn < 5000) |
27 and 28
|
| 5–5–5 |
(+)-8c |
TiCl2 |
MAO
|
1-Hexene–styrene |
>1290 |
|
|
|
|
Oligomerization, optically active (Mn < 5000) |
27 and 28
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
4-MPD
d |
57000 |
1.7 |
146 |
|
iso
|
1 × 10−5 mol of 6f, Al/Ti = 2200; 1,2-fasion |
38
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Styrene–ethylene |
|
|
|
|
|
1 × 10−6 mol of 6f, Al/Ti = 900, ethylene (1 atm); isotactic PS containing a low amount of ethylene units, mmMmmcopolymer |
39
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Styrene–propylene |
33000 |
1.9 |
171/192 |
|
iso
|
2.5 × 10−5 mol of 6f, Al/Ti = 500, propylene (1 atm); iPS-block-iPP-block-iPS copolymer |
41
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Ethylene–butadiene |
16000–64000* |
1.9–3.4 |
|
96–1332 |
|
1 × 10−6 mol of 6f, Al/Ti = 1000, 0–50 °C in toluene |
42
|
| 5–5–5 |
6f
|
TiCl2 |
MAO
|
Ethylene–4-MPDd |
4900–7100 |
1.31–2.11 |
|
128–298 |
|
1 × 10−5 mol of 6f, Al/Ti = 1200, in toluene |
43
|
| 5–5–5 |
6h
|
TiCl2 |
MAO
|
Ethylene-4-MPD
d |
10400–104000 |
1.31–2.11 |
85.3–115.6 |
60–411 |
|
1 × 10−5 mol of 6h, Al/Ti = 1200, in toluene; 4-MPD content 40–83%, Tg −37.9–2.4 °C |
43
|
| 5–5–5 |
6h
|
TiCl2 |
MAO
|
Styrene
|
|
1.05–1.19 |
|
|
iso
|
1 × 10−6 mol of 6h, Al/Ti = 1200, in toluene; living polymerization |
45
|
| 5–5–5 |
6h
|
TiCl2 |
MAO
|
Butadiene
|
|
1.28–1.77 |
|
|
|
1 × 10−5 mol of 6h, Al/Ti = 1200, in toluene; trans-1,4 selective (cis-1,4 <5%), living polymerization |
45
|
| 5–5–5 |
6h
|
TiCl2 |
MAO
|
Styrene–butadiene |
PS: 108000–171000 |
PS: 1.04–1.08 |
PS: 222.8–224.1 |
|
|
1 × 10−6 mol of 6h, Al/Ti = 1200, 25 °C in toluene; iPS-block-1,4-trans-PB copolymer |
45
|
|
PS-b-PB: 139000–201000 |
PS-b-PB: 1.07–1.11 |
PB: 40.4/82.9–42.6/85.9 |
| 5-5–5 |
9f
|
TiBn2 |
B(C6F5)3 |
1-Hexene
|
352 |
1.33 |
|
|
|
rt; TOF > 19960–20700 h−1; 2,1-regioselectivity (92–99%) |
46
|
| 5–5–5 |
19
|
TiMe2 |
B(C6F5)3 |
1-Hexene
|
352 |
1.30 |
|
|
|
rt; TOF > 19960–20700 h−1; 2,1-regioselectivity (92–99%) |
46
|
| 5–5–5 |
10
|
ZrBn2 |
B(C6F5)3 |
1-Hexene
|
224 |
1.23 |
|
|
|
TOF = 196 h−1 |
46
|
| 5–5–5 |
11
|
HfBn2 |
B(C6F5)3 |
1-Hexene
|
338 |
1.38 |
|
|
|
TOF = 3.8 h−1 |
46
|
| 6–5–6 |
16
|
ZrBn2 |
B(C6F5)3 |
1-Hexene
|
7400 |
1.6 |
|
80 |
a
|
rt |
29
|
| 6–5–6 |
17
|
ZrBn2 |
B(C6F5)3 or boratee |
1-Hexene
|
41000–120000* |
1.6–2.1 |
|
2500 |
iso
|
in benzene-hexane; exothermic, mmmm > 95% |
30
|
As investigation on active species for the olefin polymerizations mentioned above, Okuda reported NMR studies on cationic titanium benzyl species 20++·[PhCH2B(C6F5)3]− generated from 9f bearing a 5–5–5 [OSSO]-type ligand 6f by treatment with B(C6F5)3 in C6D5Br.32 The two diastereomers of 20++·[PhCH2B(C6F5)3]− were distinguishable in the 13C NMR at low temperature (−30 °C), where the signals due to TiCH2 carbons were observed in nearly equal intensity. This result showed that the ratio of both C1-cymmetric diastereomers was nearly equal and suggested that the epimerization between Δ- and Λ-forms did not occur on the NMR time scale at this temperature (Scheme 22). On the other hand, the activation of titanium dibenzyl complexes incorporating 5–6–5 [OSSO]-type ligands 3a and 3b with B(C6F5)3 in C6D5Br at −30 °C led to decomposition of paramagnetic species such as Ti(III) species.
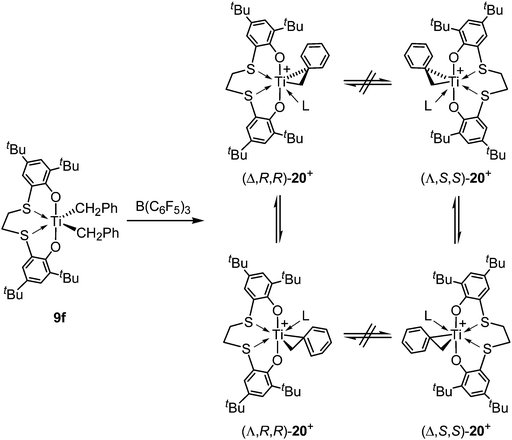 |
| | Scheme 22 Preparation of cationic titanium benzyl complex 20++ by treatment of 9f with B(C6F5)3. | |
The cationic titanium methyl complex 21++ was isolated as a Lewis-base stabilized form by Okuda et al.46 Upon treatment of the titanium dimethyl complex 19 with 1 equivalent of B(C6F5)3 in the presence of DMPE (DMPE = Me2PCH2CH2PMe2) in toluene, thermally stable (up to 60 °C) cationic species was formed as part of the ion pair 21++·[MeB(C6F5)3]− (Scheme 23). Cationic species 21++·[MeB(C6F5)3]− was fully characterized by NMR spectroscopy. X-Ray crystallography revealed that 21++·[MeB(C6F5)3]− featured separate ion pairs in the solid state and the titanium center was coordinated by two trans-oxygen and two cis-sulfur donors of helical [OSSO]-type ligand, the two phosphorus atoms of the DMPE, and a methyl group, resulting in a pentagonal bipyramidal geometry.
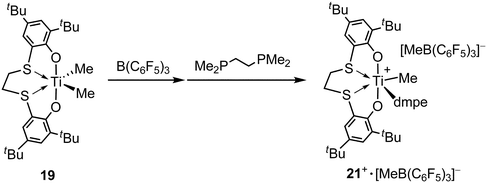 |
| | Scheme 23 Synthesis of cationic titanium methyl complex 21++ stabilized by DMPE. | |
In the case of cationic zirconium complex, the cationic zirconium benzyl complex 22++·[PhCH2B(C6F5)3]−, prepared by treatment of 17 with B(C6F5)3 in C6D6, was also observed by 1H and 19F NMR spectroscopies (Scheme 24).47 In the 19F{1H} NMR, three inequivalent 19F nuclei in the counter anion were observed at δ −166.5 (m-F), −163.8 (p-F), and −130.1 (o-F) ppm. The Δδ {|δm-F − δp-F|} value was 2.7 ppm, which indicates weak coordination between the counter anion [PhCH2B(C6F5)3]− and the zirconium cation center.48 The 1H NMR spectrum of 22++·[PhCH2B(C6F5)3]− indicated a clean formation of a single zirconium cation with the C1 symmetry. Thus, four singlets due to tert-butyl groups and three pairs of two doublets due to three benzyl groups appeared inequivalently.
 |
| | Scheme 24 Preparation of the cationic zirconium benzyl complex 22++ by treatment of 17 with B(C6F5)3. | |
Conclusion and outlook
In this review, we have presented the syntheses and olefin polymerization abilities of structurally well-defined post-metallocene initiators incorporating a new family of [OSSO]-type tetradentate dianionic ligands. These [OSSO]-type ligands are easily prepared by a few reaction steps on a gram-order scale. Group 4 metal catalysts based on compact 5–5–5 [OSSO]-type ligand 1 efficiently polymerize styrene to give isotactic polystyrene with good activity upon activation by MAO. The living isotactic polymerization of styrene was achieved with borate-activated titanium dibenzyl complex having 1e, and the first example of optically active oligostyrenes was produced by the titanium dichloro complex with enantiopure 5–5–5 [OSSO]-type ligand 2d. Furthermore, the titanium dichloro precatalyst (6f) with 1f was applied to copolymerizations of styrene/ethylene and styrene/propylene systems. In the case of 6–5–6 [OSSO]-type ligands, the combination of zirconium dibenzyl complex 17 bearing ligand 5 and (Ph3C)[B(C6F5)4] as the activator provided poly(1-hexene)s with almost perfect isospecificity in the polymerization of 1-hexene, where the activity is the highest in those of reported isospecific 1-hexene polymerization catalyzed by single-site post-metallocene complexes.22,49,50 It is anticipated that the outcomes of these olefin polymerizations with Group 4 metal complexes featured [OSSO]-type ligands will allow further improvement of [OSSO]-type ligands aiming at accomplishing stereoselective and living polymerizations involving an extremely high activity. In particular, the stereoselective and living polymerizations will lead to new block copolymerization of non-polar olefins having secondary structures.
References
- H. Sinn and W. Kaminsky, Adv. Organomet. Chem., 1980, 18, 99–149 CAS.
-
J. Scheirs and W. Kaminsky, Metallocene Based Polyolefins, Wiley & Sons, Chichester, 2000, vol. I & II Search PubMed.
-
L. S. Baugh and J.-A. M. Canich, Stereoselective Polymerization with Single-Site Catalysts, CRC Press, Boca Raton, FL, 2008 Search PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS.
- V. C. Givson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS.
- V. C. Givson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS.
- Y. Suzuki, H. Terao and T. Fujita, Bull. Chem. Soc. Jpn., 2003, 76, 1493–1517 CrossRef CAS.
- T. Matsugi and T. Fujita, Chem. Soc. Rev., 2008, 37, 1264–1277 RSC.
- H. Terao, N. Nagai and T. Fujita, J. Synthetic Org. Chem., 2008, 66, 444–457 Search PubMed.
- E. Y. Tshuva, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2000, 122, 10706–10707 CrossRef CAS.
- S. Segal, I. Goldberg and M. Kol, Organometallics, 2005, 24, 200–202 CrossRef CAS.
- A. Yeori, I. Goldberg, M. Shuster and M. Kol, J. Am. Chem. Soc., 2006, 128, 13062–13063 CrossRef CAS.
- E. Kirillov, L. Lavanant, C. Thomas, T. Roisnel, Y. Chi and J.-F. Carpentier, Chem.–Eur. J., 2007, 13, 923–935 CrossRef CAS.
- S. Gendler, A. L. Zelikoff, J. Kopilov, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2008, 130, 2144–2145 CrossRef CAS.
- A. Cohen, A. Yeori, J. Kopilov, I. Goldberg and M. Kol, Chem. Commun., 2008, 2149–2151 RSC.
- A. Cohen, J. Kopilov, I. Goldberg and M. Kol, Organometallics, 2009, 28, 1391–1405 Search PubMed.
- A. Yeori, I. Goldberg and M. Kol, Macromolecules, 2007, 40, 8521–8523 CrossRef.
- S. Segal, A. Yeori, M. Shuster, Y. Rosenberg and M. Kol, Macromolecules, 2008, 41, 1612–1617 CrossRef CAS.
- G. J. M. Meppelder, H.-T. Fan, T. P. Spaniol and J. Okuda, Organometallics, 2009, 28, 5159–5165 Search PubMed.
- T. Miyatake, K. Mizunuma, Y. Seki and M. Kakugo, Makromol. Chem. Rapid Commun., 1989, 10, 349–352 CrossRef CAS.
- T. Miyatake, K. Mizunuma and M. Kakugo, Makromol. Chem., Macromol. Symp., 1993, 66, 203–214 Search PubMed.
- J. Schaverien, A. J. van der Linden and A. G. Orpen, J. Am. Chem. Soc., 1995, 117, 3008–3021 CrossRef CAS.
- R. D. Froese, D. G. Musaev, T. Matsubara and K. Morokuma, J. Am. Chem. Soc., 1997, 119, 7190–7196 CrossRef CAS.
- R. D. Froese, D. G. Musaev and K. Morokuma, Organometallics, 1999, 18, 373–379 CrossRef CAS.
- C. Capacchione, A. Proto, H. Ebeling, R. Mülhaupt, K. Möller, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2003, 125, 4964–4965 CrossRef CAS.
- C. Capacchione, R. Manivannan, M. Barone, K. Beckerle, R. Centore, L. Oliva, A. Proto, A. Tuzi, T. P. Spaniol and J. Okuda, Organometallics, 2005, 24, 2971–2982 CrossRef CAS.
- K. Beckerle, R. Manivannan, B. Lian, G.-J. M. Meppelder, G. Raabe, T. P. Spaniol, H. Ebeling, F. Pelascini, R. Mülhaupt and J. Okuda, Angew. Chem., Int. Ed., 2007, 46, 4790–4793 CrossRef CAS.
- G.-J. M. Meppelder, K. Beckerle, R. Manivannan, B. Lian, G. Raabe, T. P. Spaniol and J. Okuda, Chem.–Asian J., 2008, 3, 1312–1323 CrossRef CAS.
- A. Cohen, A. Yeori, I. Goldberg and M. Kol, Inorg. Chem., 2007, 46, 8114–8116 CrossRef CAS.
- A. Ishii, T. Toda, N. Nakata and T. Matsuo, J. Am. Chem. Soc., 2009, 131, 13566–13567 CrossRef CAS.
- K. Beckerle, C. Capacchione, H. Ebeling, R. Manivannan, R. Mülhaupt, A. Proto, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2004, 689, 4636–4641 CrossRef CAS.
- K. Beckerle, R. Manivannan, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 3019–3026 CrossRef CAS.
- T. Toda, N. Nakata, T. Matsuo and A. Ishii, J. Organomet. Chem., 2011, 696, 1258–1261 Search PubMed.
- F. De Carlo, C. Capacchione, V. Schiavo and A. Proto, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1486–1491 CrossRef CAS.
- C. Capacchione, A. Proto and J. Okuda, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2815–2822 CrossRef CAS.
- S. Milione, C. Cuomo, C. Capacchione, C. Zannoni, A. Grassi and A. Proto, Macromolecules, 2007, 40, 5638–5643 Search PubMed.
- C. Capacchione, A. Proto, V. Venditto and J. Okuda, Macromolecules, 2003, 36, 9249–9251 CrossRef CAS.
- C. Capacchione, M. D'Acunzi, O. Motta, L. Oliva, A. Proto and J. Okuda, Macromol. Chem. Phys., 2004, 205, 370–373 CrossRef CAS.
- L. Porri and M. C. Gallazzi, Eur. Polym. J., 1966, 2, 189–198 Search PubMed.
- C. Capacchione, A. Proto, H. Ebeling, R. Mülhaupt and J. Okuda, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1908–1913 CrossRef CAS.
- C. Capacchione, F. De Carlo, C. Zannoni, J. Okuda and A. Proto, Macromolecules, 2004, 37, 8918–8922 CrossRef CAS.
- C. Capacchione, A. Avagliano and A. Proto, Macromolecules, 2008, 41, 4573–4575 CrossRef CAS.
- A. Proto, A. Avagliano, D. Saviello and C. Capacchione, Macromolecules, 2009, 42, 6981–6985 Search PubMed.
- C. Capacchione, D. Saviello, A. Avagliano and A. Proto, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4200–4206 Search PubMed.
- A. Proto, A. Avagliano, D. Saviello, R. Riccardi and C. Capacchione, Macromolecules, 2010, 43, 5919–5921 Search PubMed.
- B. Lian, K. Beckerle, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2007, 46, 8507–8510 CrossRef CAS.
-
A. Ishii, T. Toda, N. Nakata, and T. MatsuoPhosphorus, Sulfur Silicon Relat. Elem., in press Search PubMed.
- D. Horton, J. de With, A. J. van der Linden and H. van de Weg, Organometallics, 1996, 15, 2672–2674 CrossRef CAS.
- B. D. Ward, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 1668–1671 CrossRef CAS.
- P. Sudhakar and G. sundararajan, Macromol. Rapid Commun., 2005, 26, 1854–1859 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 

