DOI:
10.1039/C1PY00049G
(Paper)
Polym. Chem., 2011,
2, 1505-1512
Synthesis of heterotelechelic polymers with affinity to glutathione-S-transferase and biotin-tagged proteins by RAFT polymerization and thiol–ene reactions†
Received
28th January 2011
, Accepted 21st March 2011
First published on 15th April 2011
Abstract
α-Glutathione (GSH), ω-biotin functionalized poly(N-isopropylacrylamide) (PNIPAAm) was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization using a new R-group allyl functionalized trithiocarbonate chain transfer agent (CTA) and thiol–ene reactions. GPC and 1H NMR results indicated that the allyl group had no adverse effect on the RAFT-controlled polymerization of NIPAAm and PEG-A, and the new CTA could efficiently control the polymerizations. Employing radical thiol–ene and Michael addition reactions, heterotelechelic α-allyl, ω-carboxylic acid-PNIPAAm was first aminolyzed in the presence of maleimide-modified biotin and subsequently reacted with GSHvia radical thiol–ene addition to yield α-GSH, ω-biotin functionalized PNIPAAm. Glutathione S-transferase (GST) and streptavidin (SAv) were coupled in solution with heterofunctional PNIPAAmvia bioaffinity interactions. Separately, α-GSH, ω-biotin functionalized PNIPAAm was further shown to bind GST-tagged Rac1, a potential cancer marker, and biotin-tagged bovine serum albumin (BSA).
Introduction
Conjugation of polymers to proteins can provide proteins with new hybrid properties useful for applications in biomedicine, biotechnology, and nanotechnology.1–3 To date various well-defined protein–polymer conjugates including semitelechelic conjugates4–10 combining one protein with one polymer chain, homotelechelic conjugates11,12 combining two proteins of the same type via one polymer chain, heterotelechelic conjugates13–15 combining two different types of proteinsvia one polymer chain, and heterogenous conjugates6,16–19 combining one protein with several polymer chains have been developed. Polymers such as poly(ethylene glycol) (PEG),10,19 poly(PEG-(meth)acrylate),4,16 poly(N-(2-hydroxypropyl) methacrylamide) (PHPMA),6 poly(hydroxyethyl (meth)acrylate),5 temperature-responsive poly(N-isopropylacrylamide) (PNIPAAm)5,11–13 have been favorably used for modification of proteins.
Despite the increasing use of protein–polymer conjugates, a generic method for conjugation of polymers to different proteins has not yet been developed. Depending on the protein(s) to be conjugated, the termini of the polymers have to be functionalized according to the most proper conjugation chemistry. An alternative approach might be to utilize biological linkers having bioaffinity toward protein-tags, such as biotin and glutathione-S-transferase, widely employed in biotechnological processes.
A wide variety of biotin-tagged proteins have been expressed using recombinant protein techniques.20Biotin has a high affinity toward streptavidin (SAv)21 which is one of the most-widely used biological linkers. The high binding affinity and the presence of four biotin binding sites have favored the widespread application of SAv as a universal bio-linker. Similarly, glutathione-S-transferase (GST) tagged recombinant proteins have been commonly produced and used in biotechnological processes. GST binds glutathione (GSH) with an affinity constant Ka = 104 M−1.22 The binding between GST and GSH does not involve the free thiol residue of GSH. Considering the versatility and wide-applicability of biotin- and GST-tagged proteins, functionalization of polymer end-groups with SAv and GSH potentially provides a generic and mild route to the polymer conjugates of a wide-variety of proteins. In a recent publication, GSH-terminated poly(N-isopropylacrylamide) was shown to have selectivity against GST among other proteins such as albumin and lysozyme, and used to capture GST in solution.23
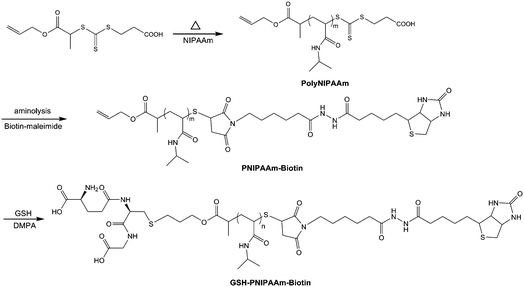
The reversible addition–fragmentation chain transfer (RAFT) polymerization is a powerful tool to the generation of well-defined heterotelechelic polymers24–30 and protein–polymer conjugates.11,13,14 Herein, we report the RAFT synthesis of heterotelechelic poly(N-isopropylacrylamide) (PNIPAAm) and its functionalization with biotin and GSH for further conjugations with SAv, and biotin- and GST-tagged proteins (Scheme 1). A new trithiocarbonate chain transfer agent (CTA) having an allyl functionalized R-group was used to generate α-allyl-PNIPAAm. Following ω-group modification of PNIPAAm by aminolysis in the presence of hexylamine, maleimide-modified-biotin was introduced to the ω-terminal of the polymerviaMichael addition. Subsequently, GSH was introduced to the α-terminal of the polymervia radicalic thiol–ene addition using a photoinitiator under UV-radiation. The bioaffinity interaction of α-GSH, ω-biotin-PNIPAAm with SAv and GST, and also with GST-tagged Rac1 protein and biotin-tagged bovine serum albumin was investigated.
 |
| | Scheme 1
RAFT polymerization of NIPAAm using a new allyl-functionalized CTA, and end-group functionalization with biotin and reduced glutathione (GSH). | |
Experimental section
Materials
The initiator, 2,2-azobisisobutyronitrile (AIBN), was recrystallized twice from methanol prior to use. N-Isopropylacrylamide (NIPAAm) (97%) was recrystallized twice from hexane prior to use (mp 64 °C). Poly(ethylene glycol) methyl ether acrylate (PEG-A) (Aldrich, Mn 480 g mol−1) was run through an alumina column to remove the inhibitor prior to use. Acetonitrile, diethyl ether, and dichloromethane (Univar, analytical grade reagent) were used as received. Biotin-maleimide, triethylamine, hexylamine, allyl alcohol, 2,2-dimethoxy-2-phenylacetophenone (DMPA), glutathione, 2,4,6-trinitrobenzene sulfonic acid (TNBSA) solution, HABA/avidin reagent, biotin labeled bovine serum albumin, glutathione S-transferase from equine liver, streptavidin from Streptomyces avidinii were purchased from Sigma-Aldrich and used without further purification. GST-tagged wild type Rac1 protein (50 kDa) was purchased from Jomar Bioscience Pty Ltd. 4–20% TGX precast gel, tris/glycine/SDS buffer, native loading buffer, Coomassie blue staining solution were purchased from Bio-Rad.
Synthesis of RAFT agent: 3-((1-(allyloxy)-1-oxopropan-2-ylthio)carbonothioylthio) propanoic acid.
The synthesis of RAFT agent was achieved in several steps as shown in Scheme S1 in the ESI†.
Synthesis of allyl 2-bromopropanoate (1).
Allyl alcohol (or prop-2-en-1-ol) (5.8 g, 0.1 mol), dichloromethane (25 mL) and triethylamine (TEA) (12.0 g, 0.12 mol) were introduced in the round bottom flask equipped by a septum. The flask is placed in ice bath, and a solution of DCM containing bromopropionic bromide (23.0 g, 0.11 mol) was added dropwise to the solution of allyl alcohol. At the end of the addition, the ice bath was removed and the solution was stirred overnight at room temperature. A white precipitate was observed and it was removed by filtration. The filtrate was washed three times with neutral water and twice with acidic water (with [HCl] = 0.1 M) to remove the trace of TEA, and finally, washed twice with neutral water. The organic phase was dried over sodium sulfate (Na2SO4) and condensed using an evaporator to remove DCM and unreacted allyl alcohol to yield a clear viscous solution (yield 90%, 24.5 g). The product was analyzed by 1H and 13C NMR spectrometry. 1H NMR (300 MHz, CDCl3, δ (ppm)): 6.05 (m, 1H, allyl group), 5.20–5.30 (m, 2H, allyl group), 4.60 (d, 2H, CH2O), 4.44 (q, 1H, CH–Br), 1.80 (d, 3H, CH3C–Br).
13C NMR (75 MHz, CDCl3, δ (ppm)): 168.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 132.2 (CH2CH), 118.4 (CH2CH), 40.8 (C–Br); 21.4 (CH3). ESI MS: 214.9 (MNa+).
O); 132.2 (CH2CH), 118.4 (CH2CH), 40.8 (C–Br); 21.4 (CH3). ESI MS: 214.9 (MNa+).
Synthesis of dithiocarboxysulfanyl propionic acid salt (2).
3-Mercaptopropionic acid (4 mL, 46 mmol) was mixed with dichloromethane (50 mL). The solution was stirred for 10 min, followed by the drop-wise addition of carbon disulfide (6 mL, 62 mmol) in an ice bath. The orange oil was stirred at ambient temperature for 5 hours. The solvent was partially evaporated under vacuum and the product was precipitated in diethyl ether to yield an orange oil. The process was repeated three times. The orange oil was separated from diethyl ether and any trace of diethyl ether was removed by a vacuum pump (yield 80%). 1H NMR confirmed the structure expected. 1H NMR (300 MHz, CDCl3, δ (ppm)): 2.9 (12H, q, N–CH2– of TEA), 3.7 (2H, t, –CH2–COO−), 2.5 (2H, t, –CH2–S–), 1.2 (18H, t, CH3 of TEA).
Synthesis of 3-((1-(allyloxy)-1-oxopropan-2-ylthio)carbonothioylthio) propanoic acid (3).
Allyl 2-bromopropanoate (1) (9.75.5 g, 0.05 mol) was dissolved in DCM (50 mL) and stirred with a 1.2-fold excess of dithiocarboxysulfanyl propionic acid salt (24.50 g, 67.0 mmol) for 16 h at room temperature. The triethylammonium bromide salt was filtered off and washed with DCM (5 mL). The product was purified by extraction (twice) with deionized water (20 mL) and three times with acidic water (pH 2). The organic phase was dried over sodium sulfate (Na2SO4) and the solvent was then evaporated. The final yellow oil was purified using a silica gel column (chloroform) (yield 65%).
1H NMR (300 MHz, CDCl3, δ (ppm)): 8.85 (br s, C(O)–OH), 5.9 (m, 1H, allyl group), 5.20–5.30 (m, 2H, allyl group), 4.85 (q, 1H, –CH–S(CS)), 4.60 (d, 2H, CH2O), 3.6 (t, 2H, –CH2–COO−), 2.5 (t, 2H, –CH2–S–), 1.60 (d, 3H, CH3C–S(CS)).
13C NMR (75 MHz, CDCl3, δ (ppm)): 224.1 (CS), 175.2 (CO, acid), 172.4 (CO, ester), 132.2 (CH2CH), 118.4 (CH2CH), 65.8 (CO–O–CH2), 46.8 (C(CH3)–S), 35.6 (CH2–S), 32.4 (CH2–CO2H), 18.4 (CH3).
RAFT polymerizations
.
Two monomers PEG-A and NIPAAm were polymerized using the new CTA. The typical procedure for polymerization of PEG-A is as follows: the CTA 1 (11.7 mg, 0.04 mmol), PEG-A monomer (960 mg, 2 mmol), AIBN (1.3 mg, 0.008 mmol) were dissolved in acetonitrile (2.1 mL). Following the sealing of the vial with rubber septum, the polymerization solution was purged with nitrogen for 30 min in an ice bath. The final concentration of the monomer, CTA, and the initiator in the polymerization medium was 1, 0.02 and 4 × 10−3 M, respectively ([monomer]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA]0:[initiator]0 50:1.0:0.2). The solution was placed in an oil bath at 60 °C. Aliquots (0.1 mL) were taken at predetermined time intervals and quenched via rapid cooling and exposure to oxygen. These samples were directly analyzed by 1H NMR and THF GPC to determine the monomer conversion and the molecular weight, respectively. The polymer was concentrated by partial evaporation of acetonitrile, then dialyzed against water for 2 days to remove nonreacted monomer and the CTA. After freeze-drying, a yellow product was obtained. The samples were further analyzed by GPC and 1H NMR. The polymerization of PNIPAAm follows the same procedure as described above, except the molar feed ratio [NIPAAm]0/[CTA]0/[AIBN]0 = 50/1/0.2, [NIPAAm] = 1.0 mol L−1.
:[CTA]0:[initiator]0 50:1.0:0.2). The solution was placed in an oil bath at 60 °C. Aliquots (0.1 mL) were taken at predetermined time intervals and quenched via rapid cooling and exposure to oxygen. These samples were directly analyzed by 1H NMR and THF GPC to determine the monomer conversion and the molecular weight, respectively. The polymer was concentrated by partial evaporation of acetonitrile, then dialyzed against water for 2 days to remove nonreacted monomer and the CTA. After freeze-drying, a yellow product was obtained. The samples were further analyzed by GPC and 1H NMR. The polymerization of PNIPAAm follows the same procedure as described above, except the molar feed ratio [NIPAAm]0/[CTA]0/[AIBN]0 = 50/1/0.2, [NIPAAm] = 1.0 mol L−1.
Synthesis of ω-biotin poly(NIPAAm) (PNIPAAm-biotin).
DMSO (1 mL), PNIPAAm (8 × 10−3 mmol) and biotin-maleimide (0.02 mmol) were mixed together, and the solution was purged with nitrogen for 30 min. Hexylamine (38 μmol) and TEA (14.4 μmol) were then added using a syringe. The mixture was stirred overnight at room temperature. The product was purified by dialyzing in water for 3 days. After freeze-drying, a white powder was obtained.
Synthesis of α-GSH, ω-biotin poly(NIPAAm) (GSH-PNIPAAm-biotin).
In 1 mL of DMSO and water (50:50 vol%), PNIPAAm-biotin (7 × 10−3 mmol) was dissolved. Glutathione (GSH) and then the photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPA), were added. [GSH]/[polymer]/[photoinitiator] mol ratio was 120:1:0.3. Following the sealing of the vial with rubber septum, the solution was purged with nitrogen for 30 min. Under irradiation with a 365 nm UV lamp (6 watt), the reaction was allowed to proceed for 2 h. The product was purified by dialyzing in water for 3 days. The final product was obtained by freeze-drying.
Determination of biotin in the polymer by HABA/avidin reagent.
The purchased HABA/avidin reagent lyophilized powder was reconstituted by adding 10 mL deionized water. In a 1 mL cuvette, HABA/avidin reagent (900 μL) was added, and the absorbance at 500 nm was recorded. 100 μL of sample was then added, mixed by pipette, and the A500 was measured. Considering that the assay is based on the binding of the dye HABA to avidin and the ability of biotin to displace the dye in stoichiometric proportions, this displacement of dye is accompanied by a change in the absorbance at A500 which has a known extinction coefficient (34000 M−1 cm−1).13 Accordingly, from the following equation, the concentration of biotin in the sample was calculated.
| HABA/avidin+sample(500 nm) |
where 0.9 = dilution factor of HABA/avidin upon addition of sample; 34 = extinction coefficient at 500 nm (in mM−1 cm−1); 10 = dilution factor of the sample in the cuvette.
Determination of glutathione conjugated to the polymer by TNBSA reagent.
Polymer (50 μg mL−1) was dissolved in 0.1 M sodium bicarbonate buffer (pH 8.5). The supplied 5% TNBSA solution was diluted 1000-fold in 0.1 M sodium bicarbonate buffer (pH 8.5). The diluted TNBSA solution (0.25 mL) was added to 0.4 mL of sample solution, and incubated at 37 °C for 2 hours. Then 10% SDS (0.5 mL) and 1 N HCl (0.25 mL) were added to each sample to stop and stabilize reaction. The absorbance of the solution was measured at 335 nm. To determine the concentration of amine, a standard glycine solution standard was given in the same procedure. By comparison to the standard, the content of GSH in the polymer was obtained. As a control, PNIPAAm without GSH was also tested using the same protocol.
Instrumental analyses
Gel Permeation Chromatography (GPC).
GPC analyses were performed using tetrahydrofuran (THF) or N,N-dimethylacetamide (DMAC)/0.05 mol% lithium bromide (LiBr) as the mobile phase. The GPC Shimadzu modular system comprised of an LC-10ATVP Shimadzu solvent delivery system, a SIL-10ADVP Shimadzu autoinjector, a column set that consisted of a Phenomonex 5.0 μm bead size guard column and four 5.0 μm Phenomonex Phenogel columns (500, 103, 104, and 106 Å for the GPC using THF as a mobile phase, and 500, 103, 104, and 105 Å for the GPC using DMAC as a mobile phase), and a differential refractive-index detector. The temperature was kept constant at 40 °C using a CTO-10AC VP Shimadzu column oven. The columns were calibrated with commercial linear polystyrene standards ranging from 500 to 106 g mol−1.
Gel electrophoresis
.
Gel electrophoresis was performed using 4–20% polyacrylamide gels. Samples were loaded with 5× native loading buffer, and run at a constant voltage of 200 V for 30 minutes using 1× TG/SDS buffer (tris-glycine, pH 8.3). The gel was stained with Bio-Safe Coomassie for 30 minutes.
Results and discussion
A new CTA (3 of Scheme S1†) containing an R-group functionalized with allyl group was synthesized. ESI-MS and 1H NMR analyses confirmed the successful synthesis of the RAFT agent (Fig. S1 in the ESI†) with a purity higher than 95%. This new CTA was tested in the polymerization of both NIPAAm and PEG-A monomers (Fig. S2 in the ESI† and Fig. 1) to show the utility of the new RAFT agent with both acrylate and acrylamide monomers. Fig. 1A shows the evolution of monomer conversion versus time. A short inhibition period (approx. 20–25 min) was observed. This was attributed to a range of causes from slow fragmentation31 to initialization32 and impurities, and the trace of oxygen.33,34 Specific inhibition/retardation phenomena with trithiocarbonates have been observed previously where trithiocarbonates were found to be unsuitable RAFT agents for the study of termination rate coefficients.33 This problem of retardation/inhibition with trithiocarbonates in these earlier careful kinetic studies was attributed to additional radical loss pathways (unspecified). Post-inhibition, both polymerizations (PEG-A and NIPAAm) proceeded in an identical fashion, consistent with the known traits of living radical polymerizations. The linear plot of ln ([M]0/[M]) versus time (min) indicates that the concentration of radical was constant during polymerization. NIPAAm polymerized faster than PEG-A. The evolution of molecular weights, characterized by GPC and by NMR analyses, was linear with monomer conversion. As the reaction proceeded, the molecular weight distributions remained narrow (PDI ≤ 1.20), indicating a well-controlled polymerization. After removing residual monomer and impurities by dialysis against water, the polymers were analyzed by 1H NMR and GPC.
![RAFT polymerization of NIPAAm in acetonitrile at 60 °C. [NIPAAm]0/[CTA]0/[AIBN]0 = 50/1/0.2, [NIPAAm] = 1.0 mol L−1; (A) GPC traces, (B) polymerization kinetic plots: monomer conversion versus time and ln ([M]0/[M] versus time); (C) evolution of the number average molecular weight (Mn) determined by (blue triangle) GPC and (black square) by NMR and (blue diamond) polydispersity (PDI) versus conversion.](/image/article/2011/PY/c1py00049g/c1py00049g-f1.gif) |
| | Fig. 1
RAFT polymerization of NIPAAm in acetonitrile at 60 °C. [NIPAAm]0/[CTA]0/[AIBN]0 = 50/1/0.2, [NIPAAm] = 1.0 mol L−1; (A) GPC traces, (B) polymerization kinetic plots: monomer conversion versus time and ln ([M]0/[M] versus time); (C) evolution of the number average molecular weight (Mn) determined by (blue triangle) GPC and (black square) by NMR and (blue diamond) polydispersity (PDI) versus conversion. | |
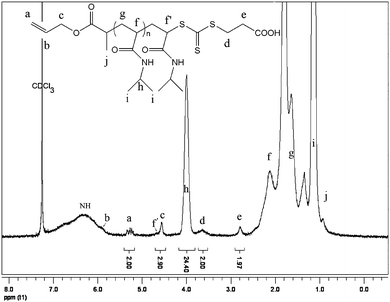
The 1H NMR spectrum of the purified PNIPAAm (Mn by GPC = 6100 g mol−1, PDI = 1.12, conversion = 53%) is shown in Fig. 2. The presence of the allylic bond was confirmed by the signals at 5.95 and 5.3 ppm by 1H NMR, while the signal of Zgroup of the RAFT agent appeared at 3.6 and 2.8 ppm attributed to –CH2CO2H and –S–CH2, respectively. The signal of CH–S appeared overlapped with the signal of O–CH2 at 4.6 ppm. The functionality of allylic bond was measured during the polymerization by 1H NMR, and calculated by the following equation: fallyl group = (I5.3 ppm/2)/(I4.6 ppm/3), with I5.3 ppm and I4.6 ppm represent the integration of the signal at 5.3 ppm and at 4.6 ppm, respectively. The functionality of allyl group stayed constant (close to 1) during the polymerization (Fig. S3, ESI†) whatever the monomer conversion was. This result indicates that the allyl group does not copolymerize to a detectable extent. Furthermore, the low PDI throughout the polymerization also indicated the absence of chain–chain coupling/branching reactions due to the copolymerization of allyl end group.
The molecular weight was also calculated by NMR using the following equation: Mn,by NMR = I4.0 ppm/(I5.3 ppm/2) × MwNIPAAm + MwRAFT, where I4.0 ppm, I5.3 ppm, MwNIPAAm and MwRAFT correspond to the integral of signal at 4.0 ppm and 5.3 ppm, molar masses of NIPAAm and RAFT agent, respectively. The values determined by NMR are in good agreement with the theoretical values calculated by the traditional equation: Mn, theoretical = [M]0/[RAFT]0 × α × MwNIPAAm + MwRAFT, where [M]0, [RAFT]0, α, MwNIPAAm and MwRAFT correspond to monomer concentration, RAFT agent concentration, monomer conversion, molar masses of NIPAAm and RAFT agent, respectively. The difference between the molecular weight values obtained by NMR and GPC was attributed to the fact that a polystyrene standard was used for the GPC calibration.
Synthesis of α-GSH, ω-biotin-PNIPAAm by thiol–ene reaction
Thiol–ene chemistry, i.e. the addition of thiols with a variety of non-activated carbon–carbon double bonds, has been described in numerous publications.34–49 The demonstrated efficiency of thiol–ene click chemistry has been widely used in polymer functionalization and conjugations.29,30,34–49 In this study, thiol–ene reactions have been employed to functionalize PNIPAAm with α-GSH, ω-biotin termini.
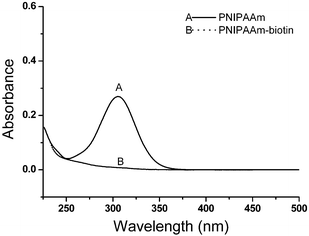
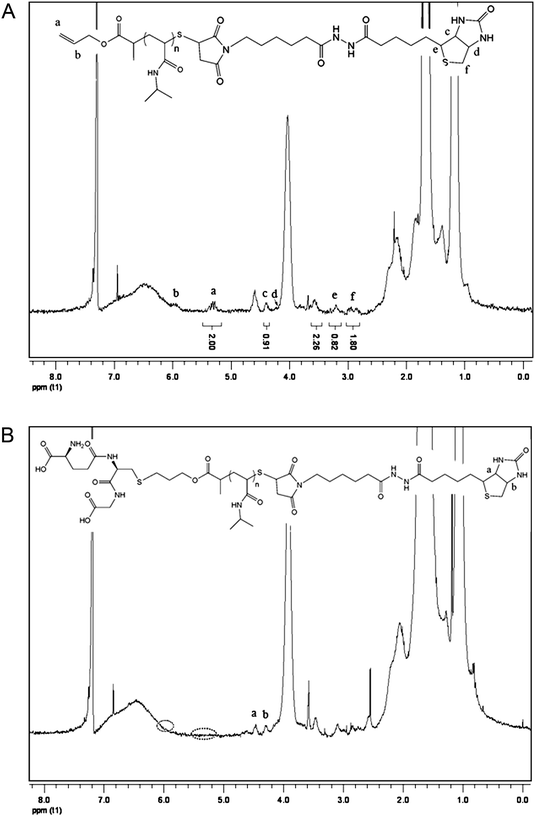
The functionalization of PNIPAAm was performed in two steps: first maleimide-functionalized biotin was reacted with PNIPAAmvia one-pot aminolysis and Michael addition reactions.29,30 During the modification, the characteristic yellow color of the polymer solution disappeared, which was consistent with the cleavage of the thiocarbonylthio end group. UV-visible spectrometry analysis of the purified polymers after the modification with biotin confirmed the disappearance of the CS band at 305 nm. The UV-visible spectra of PNIPAAm before and after biotin modification are shown in Fig. 3. Biotin conjugation was confirmed by 1H NMR analysis of the purified polymer as shown in Fig. 4A. The appearance of the new signals at 4.36 and 4.2 ppm attributed to the characteristic signals of biotin (–CH protons) (Fig. S4, in the ESI†) showed the attachment of biotin to the polymer. Additionally, the allyl end group signals (5.9 and 5.3 ppm) were still present. By calculating integration ratio between the specific peak of biotin at 4.36 ppm and allyl group at 5.3 ppm, it was confirmed that more than 90% of PNIPAAm chains were conjugated with biotin. To exactly determine the content of the biotin in the polymer, HABA/avidin reagent assay was used which was based on the binding of the dye HABA to avidin and the ability of biotin to displace the dye in stoichiometric proportions. The results indicated that 91.5% of the PNIPAAm chains were modified by biotin (Fig. S5, in the ESI†). Furthermore, GPC chromatograms that were taken before and after the biotin modification step (Fig. 5) showed that there was no significant change in the molecular weight distribution of the polymer. The monomodal distribution was retained after modification with biotin, indicating that no side reactions such as the polymer–polymer disulfide coupling occurred and the end group modification proceeded quantitatively as intended.
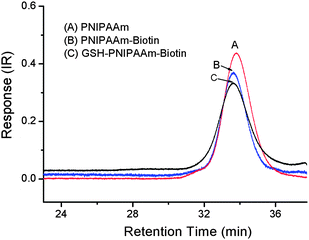 |
| | Fig. 5
GPC traces of PNIPAAm (A), PNIPAAm-biotin (B) and GSH-PNIPAAm-biotin (C), mobile phase: DMAc. | |
Following the successful synthesis of ω-biotin-functionalized PNIPAAm, the polymer was further functionalized with GSH. A radical thiol–ene addition was performed using a feed ratio [GSH]/[polymer]/[photoinitiator] of 120:1:0.3 under UV light at 365 nm. The 1H NMR spectrum of the purified polymer after modification is shown in Fig. 4B. Due to overlapping with other signals, the signal of glutathione protons could not been observed clearly. However, it was observed that the allyl proton signals at 5.9 and 5.3 ppm completely disappeared after reaction. Moreover, the GPC chromatogram of the polymer (Fig. 5, trace C) showed no shoulder of higher molecular weight, indicating that no polymer–polymer coupling occurred due to the polymerization of the allyl groups. Combination of NMR and GPC results indicated the success of α-glutathione modification of the polymer. To further confirm this conclusion and determine the level of the GSH in the polymer, a well-established TNBSA assay was performed for estimation of amine group content of the polymer. According to this assay, 91% of the polymer was conjugated with an amine compound, i.e.glutathione, determined based on the calibration curve built using a standard glycine solution (Fig. S6 in the ESI†).
Bioaffinity interactions of α-GSH, ω-biotin-PNIPAAm with streptavidin and glutathione S-transferase
Glutathione-S-transferase (GST, Mw = 25.5 kDa) or streptavidin (Mw = 60 kDa) was conjugated to α-GSH, ω-biotin-PNIPAAm (Mn = 8120 by GPC, PDI = 1.16). The conjugation was analyzed by native poly(acrylamide) gel electrophoresis (PAGE) (Fig. 6). The increase in the molecular weight of GST upon incubation with the polymer was clear on the gel (lanes 1 and 2). A control experiment (Fig. S7 in the ESI†) performed by incubating non-GSH-modified PNIPAAm with GST under the same conditions revealed that the molecular weight of GST was not affected by the presence of non-modified PNIPAAm and there was no interaction between the polymer and the protein. From these data it was clear that the GSH-modification enabled the conjugation of the α-GSH, ω-biotin-PNIPAAm to GST, as expected. The binding of SAv to α-GSH, ω-biotin-PNIPAAm was also clear with the formation of a higher molecular weight band with respect to SAv (Fig. 6, lanes 5 and 6). The relatively longer smear in lane 6 compared to the smear of SAv only indicated the formation of the conjugate with larger polydispersity. This might have resulted from the combination of the following two effects: (i) the binding of varying number of polymer chains to SAv due to the steric hindrance of the polymer chains already bound with SAv50 and (ii) the binding of the polymer chains with varying chain lengths due to the polydispersity of the polymer, PDI = 1.16. Within this study, the binding experiments were conducted using only one ratio of α-GSH, ω-biotin-PNIPAAm:SAv molar ratio (50:1). The examination of varying molar ratios should yield valuable information and be the focus of the future studies
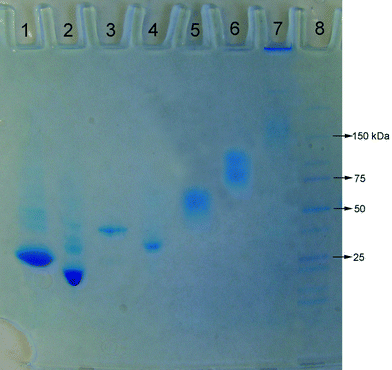 |
| | Fig. 6
Polyacrylamide gel electrophoresis of the α-GSH, ω-biotin-PNIPAAm (Mn = 8120 by GPC, PDI = 1.16) complexation with proteins. Lane 1: GST+ α-GSH, ω-biotin-PNIPAAm (GST:polymer = 1:50 mol:mol), lane 2: GST, lane 3: GST-tagged Rac1 + α-GSH, ω-biotin-PNIPAAm (GST-tagged Rac1:polymer = 1:50 mol:mol), lane 4: GST-tagged Rac1, lane 5: streptavidin, lane 6: α-GSH, ω-biotin-PNIPAAm + streptavidin (SAv:polymer = 1:50 mol:mol), lane 7: α-GSH, ω-biotin-PNIPAAm + streptavidin + biotin-tagged BSA (SAv:polymer:biotin-tagged BSA = 1:3:1), and lane 8: dual color protein marker. | |
The general applicability of α-GSH, ω-biotin-PNIPAAm to bind with different proteins having GST- or biotin-tag was tested using a potential cancer marker, namely GST-tagged Rac1 protein (Mw = ∼46 kDa), and biotin-tagged BSA (Mw = ∼67 kDa). Firstly, instead of GST, GST-tagged Rac1 protein was incubated with α-GSH, ω-biotin-PNIPAAm under the same conditions used for incubation with GST. The formation of the affinity conjugate, α-GST-tagged Rac1, ω-biotin-PNIPAAm, was clear on the gel (Fig. 6, lanes 3 and 4). Similarly, α-GSH, ω-biotin-PNIPAAm after conjugation with SAv (α-GSH, ω-SAv–PNIPAAm) was incubated with biotin-tagged BSA (10–18 moles biotin per BSA). The formation of a high molecular weight conjugate (≥150 kDa) was observed by the appearance of a retarded band at approx. 150 kDa and mostly localizing in the well (Fig. 6, lane 7). The presence of such high molecular weight structure indicated the formation of a crosslinked structure constituted by the conjugation of one biotin-tagged BSA with multiple copies of SAv-linked polymer molecule as the biotin-tagged BSA contained 10–18 moles biotin per BSA. It is anticipated that the crosslinking can be avoided if the biotin-tagging of BSA is stoichiometric.
Conclusion
The successful synthesis of a α-GSH, ω-biotin-functionalized polymer was described. The synthesis featured the use of a new CTA with an allyl-functionalized R group. Two sorts of thiol–ene reactions were employed to further modify the polymer. By simultaneous aminolysis and nucleophilic thiol–ene reaction (Michael addition), the ω-biotin was introduced to the polymer. During the polymerization and nucleophilic thiol–ene reaction, the α-allyl functionality was maintained. Glutathione was then incorporated to the α-terminal by radical thiol–ene reaction. It was further shown that α-GSH, ω-biotin-functionalized polymer can be conjugated efficiently with GST or a GST-fused protein. Lastly, the polymer, after modification with SAV, was coupled with a biotin-tagged protein. In summary, considering the versatility and wide-applicability of biotin- and GST-tagged proteins, functionalization of polymer end-groups with SAv and GSH potentially provides a generic and mild route to the polymer conjugates of a wide-variety of proteins. Such polymers would potentially be useful as molecular adapters in drug delivery and diagnostic applications.
References
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS.
- C. Boyer, X. Huang, M. Whittaker, V. Bulmus and T. Davis, Soft Matter, 2011, 7, 1599 RSC.
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Q. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS.
- L. Tao, J. Q. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS.
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 314 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860 CrossRef CAS.
- G. Pasut, A. Mero, F. Caboi, S. Scaramuzza, L. Sollai and F. Veronese, Bioconjugate Chem., 2008, 19, 2427 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148 RSC.
- C. Boyer, J. Q. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641 CrossRef CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238 CrossRef CAS.
- J. Q. Liu, H. Y. Liu, V. Bulmus, L. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399 CrossRef CAS.
- L. Tao, J. Q. Liu, J. T. Xu and T. P. Davis, Chem. Commun., 2009, 6560 RSC.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028 CrossRef CAS.
- G. P. Vlasov, Biopolymers, 2005, 80, 589.
- F. Wurm, J. Klos, H. Ra der and H. Frey, J. Am. Chem. Soc., 2009, 131, 7954 CrossRef.
- D. Luo, M. Geng, B. Schultes, J. Ma, D. Xu, N. Hamza, W. Qi, A. Noujaim and R. Madiyalakan, J. Biotechnol., 1998, 65, 225 CrossRef CAS.
- P. C. Weber, D. H. Ohlendorf, J. J. Wendoloski and F. R. Salemme, Science, 1989, 243, 85 CrossRef CAS.
- C. Dsilva, Biochem. J., 1990, 271, 161 CAS.
- C. W. Chang and H. D. Maynard, Macromol. Rapid Commun., 2010, 31, 1691 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev. (Washington, DC, U. S.), 2009, 109, 5402 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- C. Boyer, J. Q. Liu, L. J. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207 CrossRef.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493 CrossRef.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300 CrossRef CAS.
- J. B. McLeary, M. P. Tonge and B. Klumperman, Macromol. Rapid Commun., 2006, 27, 1233 CrossRef CAS.
- A. Theis, A. Feldermann, N. Charton, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Macromolecules, 2005, 38, 2595 CrossRef CAS.
- A. Favier, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Macromol. Chem. Phys., 2004, 205, 925 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev. (Washington, DC, U. S.), 2009, 109, 5620 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344 CrossRef CAS.
- J. T. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2010, 43, 20 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef CAS.
- C. Hoyle, A. Lowe and C. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- A. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- L. J. Wong, S. Sevimli, H. M. Zareie, T. P. Davis and V. Bulmus, Macromolecules, 2010, 43, 5365 CrossRef CAS.
- L. J. Wong, M. Kavallaris and V. Bulmus, Polym. Chem., 2011, 2, 385 RSC.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699 CrossRef CAS.
- M. Li, P. De, H. M Li and B. S. Sumerlin, Polym. Chem., 2010, 1(6), 854 RSC.
- C. Lavigueur, J. G. Garcia, L. Hendriks, R. Hoogenboom, J. J. L. M. Cornelissen and R. J. M. Nolte, Polym. Chem., 2011, 2, 333 RSC.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Mueller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00049g |
| ‡ X.H. and C.B. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2011 |