Macromolecular thiolysis of oxiranes: end-group modification of RAFT prepared homopolymers†
M. Alyse
Harvison
,
Thomas P.
Davis
and
Andrew B.
Lowe
*
Centre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, University of New South Wales, Kensington 2052, Sydney, Australia. E-mail: a.lowe@unsw.edu.au; Fax: +61 2 9385 5966; Tel: +61 2 9385 6031
First published on 25th March 2011
Abstract
Chain end modification of low molecular weight, RAFT-prepared polystyrene and poly(N,N-diethylacrylamide) by reaction with a range of small molecule epoxides is described. Two different routes are examined—initial dithioester end-group cleavage yielding the thiol-terminated polymer followed by catalytic thiolysis of the oxiranes and a one-pot procedure involving hydride cleavage of the dithioester end groups in the presence of an epoxide. High degrees of functionalization (>80%) are observed via the first route in the presence of ZnCl2 with molecular weight distributions remaining unimodal and narrow. However, with DBU as catalyst significant amounts of coupled species are observed. In contrast, the one-pot route, at least with poly(N,N-diethylacrylamide), resulted in essentially quantitative degrees of functionalization as evidenced by 1H NMR spectroscopy and qualitatively by FTIR spectroscopy. The effect of the newly introduced alcohol-functional end groups on the lower critical solution temperature of poly(N,N-diethylacrylamide) is demonstrated with cloud points tunable from ca. 27 to 47 °C.
Introduction
Reversible addition–fragmentation chain transfer (RAFT) polymerization is a controlled radical polymerization process mediated by thiocarbonylthio (TCT) compounds, such as dithioesters and trithiocarbonates.1–9 Such functionality remains incorporated in the resulting (co)polymers and is located at the ω-terminus in the case of typical monofunctional RAFT agents. Indeed, retention of the TCT species is crucial for the successful formation of more complex architectures such as block copolymers. However, (co)polymers prepared by RAFT are coloured due to the presence of the TCT end-groups but can be readily removed under a range of conditions yielding (co)polymers with various new end-group functionalities.10,11 Of particular interest is the cleavage of the TCT end-groups to yield thiol- or thiolate-terminal (co)polymers and this can be accomplished under a range of conditions including aminolysis,12,13 hydrazinolysis14 and hydride reduction.15,16 This is an attractive transformation since the thiol/thiolate species are able to undergo a range of facile, often quantitative, further chemical reactions.17In 2001 Kolb et al.18 highlighted an approach to the simplified synthesis/functionalisation of complex (macro)molecules they termed click chemistry.19–23 It refers to a group of chemical reactions that proceed with certain desirable features and includes Diels–Alder, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond additions, SN chemistry with an emphasis on ring-opening reactions of strained cyclic substrates and non-Aldol CO addition reactions.18 Of these, the Cu(I)-catalyzed reaction between an alkyne and azide has been the most widely exploited although it suffers from the need to use a metal-catalyst as well as potentially dangerous azides. This has led, in part, to the identification and application of alternative ‘click’ reactions.24,25 Of relevance here is the recognition of a range of thiol-based reactions26 that meet all, or most, of the criteria to be accurately designated as click reactions. These include the thiol–ene (including thiol–Michael),27–42thiol–yne,32,33,43–48thiol–isocyanate49–51 and thiol–halo52–55 reactions. Indeed, all of these chemistries have been successfully applied as a means of modifying thiol-terminated, RAFT-prepared, (co)polymers. Interestingly, of the click reactions noted above the modification of end-groups in RAFT-prepared (co)polymers via the thiolysis of strained cyclic substrates, such as oxiranes or aziridines, has not, to the best of our knowledge, been reported, even though the reaction between a thiol and epoxide are well known to proceed to high conversion under a range of conditions with, in some instances, extremely high degrees of regioselectivity.56–59 Herein we describe our preliminary observations regarding the macromolecular thiolysis of a range of commercially available epoxidesvia two different routes. We show that such chemistry can be highly efficient and offers a means of tuning the aqueous solution properties of hydrophilic homopolymers.
C bond additions, SN chemistry with an emphasis on ring-opening reactions of strained cyclic substrates and non-Aldol CO addition reactions.18 Of these, the Cu(I)-catalyzed reaction between an alkyne and azide has been the most widely exploited although it suffers from the need to use a metal-catalyst as well as potentially dangerous azides. This has led, in part, to the identification and application of alternative ‘click’ reactions.24,25 Of relevance here is the recognition of a range of thiol-based reactions26 that meet all, or most, of the criteria to be accurately designated as click reactions. These include the thiol–ene (including thiol–Michael),27–42thiol–yne,32,33,43–48thiol–isocyanate49–51 and thiol–halo52–55 reactions. Indeed, all of these chemistries have been successfully applied as a means of modifying thiol-terminated, RAFT-prepared, (co)polymers. Interestingly, of the click reactions noted above the modification of end-groups in RAFT-prepared (co)polymers via the thiolysis of strained cyclic substrates, such as oxiranes or aziridines, has not, to the best of our knowledge, been reported, even though the reaction between a thiol and epoxide are well known to proceed to high conversion under a range of conditions with, in some instances, extremely high degrees of regioselectivity.56–59 Herein we describe our preliminary observations regarding the macromolecular thiolysis of a range of commercially available epoxidesvia two different routes. We show that such chemistry can be highly efficient and offers a means of tuning the aqueous solution properties of hydrophilic homopolymers.
Experimental
All reagents were purchased from the Aldrich Chemical Company at the highest available purity and used as received unless noted otherwise. Styrene (S) was purified by passage through a basic alumina column and stored at 5 °C until needed. N,N-Diethylacrylamide was purchased from Polysciences Inc. and purified by passage over basic alumina prior to being stored at 5 °C until needed. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized twice from methanol and stored at −24 °C until needed. 1-Cyano-1-methylethyl dithiobenzoate (CPDB) was prepared according to a literature procedure.1Homopolymerization of styrene
Styrene (30.0 g, 0.28 mol) and CPDB (0.95 g, 4.3 mmol) were mixed in a round-bottomed flask equipped with a magnetic stir bar and the vessel sealed with a rubber septum. The resulting homogeneous solution was stirred at room temperature while being purged with N2 for a period of 1 h. The flask was then immersed in a preheated oil bath at 110 °C and polymerization allowed to proceed for 16 h. The polymerization was quenched by exposure to air while cooling the flask with ice. THF (10.0 mL) was then added to the flask to dilute the monomer/polymer mixture. The polymer was isolated by precipitation (twice) into methanol (900 mL) followed by Buchner filtration and drying in vacuo, yielding 11.92 g of polystyrene (PS).Homopolymerization of N,N-diethylacrylamide
N,N-Diethylacrylamide (13.3 g, 104 mmol), CPDB (0.582 g, 2.66 mmol) and AIBN (76 mg, 0.463 mmol) were mixed in a round-bottomed flask equipped with a magnetic stir bar. The resulting homogeneous solution was stirred at room temperature while being purged with N2 for a period of 1 h. The flask was then immersed in a preheated oil bath at 80 °C, for 2.5 h. The polymerization was quenched by exposure to air while cooling the flask with ice. THF (8.0 mL) was then added to the flask to dilute the monomer/polymer mixture. The polymer was isolated by precipitation (twice) into hexanes (600 mL) followed by Buchner filtration and drying in vacuo, yielding 8.7 g of poly(N,N-diethylacrylamide).Hydrazine cleavage of thiocarbonylthio end-groups in polystyrene
PS (3.1 g, 0.61 mmol) and dichloromethane were added to a round-bottomed flask equipped with a magnetic stir bar. To this solution was added hydrazine (0.155 mL, 3.1 mmol) and dimethylphenylphosphine (Me2PPh) (88.2 µl, 0.61 mmol), and the solution purged with N2 for 30 min and subsequently allowed to stir overnight at room temperature. The solution was diluted with 10 mL of dichloromethane then precipitated into methanol (900 mL). The fine precipitate was isolated by centrifugation and dried in vacuo overnight.One-pot hydride end-group cleavage/macromolecular thiolysis
Poly(N,N-diethylacrylamide) (0.2 g, 36.4 µmol), NaBH4 (2.7 mg, 73 µmol), Me2PPh (5.1 µl, 36.4 µmol), and 1,2-epoxy-5-hexene (205 µl, 1.8 mmol) were added to a vial equipped with a magnetic stir bar. To this was added THF (1 mL). The solution was purged with N2 for 2 minutes then allowed to stir for 72 hours at room temperature. The solution was diluted with 1 mL THF then precipitated (twice) into hexane (30 mL). The precipitate was isolated by centrifugation and dried in vacuo overnight.Reaction of thiol-terminated polystyrene (PS–SH) with epoxides
A typical procedure for the reaction between PS–SH and an epoxide using DBU is as follows:PS–SH (0.2 g, 36.4 µmol), Me2PPh (10 mol equiv based on thiol end groups) and DCM (1.0 mL) were added to a 5.0 mL vial equipped with a magnetic stir bar. The vial was capped with a rubber septum, and the solution purged with N2 for several minutes. The septum was opened and the target epoxide (50 mol equiv based on thiol end groups) and DBU (0.6 mol% based on thiol end groups) were added to the solution. The homogeneous solution was immersed in an oil bath preheated to 60 °C and allowed to react for 48 h. The product was isolated by precipitation (twice) into methanol (13.0 mL), followed by centrifugation and drying in vacuo overnight.
A typical procedure for the reaction between PS–SH and an epoxide in the presence of ZnCl2 is as follows:
PS–SH (0.2 g, 36.4 µmol), Me2PPh (1 mol equiv based on thiol end groups (5.1 µl)), 2-(but-3-en-1-yl)oxirane (205 µl, 36.4 µmol), ZnCl2 (36.8 mg, 15 mol% based on epoxide) and DMF (1.0 mL) were added to a 5.0 mL vial equipped with a magnetic stir bar. After complete dissolution the solution was purged with N2 for several minutes and then immersed in a preheated oil bath at 60 °C and allowed to react for 4 days. The solution was then precipitated into methanol (twice), centrifuged and the solid product dried in vacuo overnight.
Instrumentation and equipment
Molecular weight distributions were determined by size-exclusion chromatography (SEC) using a Shimadzu modular system, comprising an auto-injector, a Polymer Laboratories 5.0 µm bead-size guard column (50 × 7.5 mm), four linear PL columns (105, 104, 103 and 500 Å), and a differential refractive index detector. The mobile phase was tetrahydrofuran (THF) at a temperature of 40 °C and a flow rate of 1 mL min−1. The system was calibrated using low polydispersity polystyrene standards with molecular weights ranging from 162 to 2 × 106 g mol−1. 1H and 13C NMR spectra were recorded on a Bruker ACF300, in CDCl3 unless noted otherwise, operating at 300.17 MHz for 1H and 75.48 MHz for 13C. UV-Vis spectra were recorded on a CARY 300 spectrophotometer equipped with a temperature controller. FTIR spectra were recorded on a ThermoScientific Nicolet 5700 FTIR spectrometer.Results and discussion
Precursor homopolymers of polystyrene (PS–S(CS)Ph) and poly(N,N-diethylacrylamide) (PDEAM–S(CS)Ph) were prepared by RAFT under bulk conditions with 1-cyano-1-methylethyl dithiobenzoate (CPDB) as the RAFT chain transfer agent. In the case of S, polymerization was initiated under purely thermal conditions while for DEAM homopolymerization primary radicals were generated from the thermal decomposition of AIBN, Scheme 1. In both instances polymerization proceeded smoothly yielding homopolymers with SEC-measured molecular weights (Mn) and polydispersity indices (Mw/Mn) of 3540 (1.07) for PS–S(CS)Ph and 1790 (1.06) for PDEAM–S(CS)Ph. Absolute molecular weights were also determined by 1H NMR spectroscopy. As an example, Fig. 1 shows the 1H NMR spectrum for the precursor S homopolymer recorded in CDCl3. The signal highlighted and centered around ∼δ 4.9 ppm is attributed to the methine hydrogen on the carbon directly bonded to the TCT end-group. A ratio of the integral of this signal with that of the aromatics associated with the main chain yields a calculated average degree of polymerization (DPn) of 49. Similar end-group analysis for the PDEAM–S(CS)Ph homopolymer gave a calculated DPn of 50.
 | ||
| Scheme 1 . General polymerization outline and reaction sequences examined for the two-step (Route 1) and one-pot (Route 2) macromolecular thiolyses of small molecule epoxides. | ||
 | ||
| Fig. 1
1H NMR spectrum, recorded in CDCl3, of the parent PS–S(CS)Ph homopolymer. | ||
With the parent homopolymers in-hand two different routes were examined for effecting macromolecular thiolysis of a series of epoxides. Route 1, Scheme 1, involved initial cleavage of the TCT end-groupsvia treatment with hydrazine. Hydrazine has recently been shown to effect rapid aminolysis of such RAFT end-groups while also serving as an anti-oxidant helping to prevent/minimize disulfide formation from the aerial oxidation of the formed macromolecular thiols.14 Following formation and isolation of the thiol-terminated PS (PS–SH) this macromolecular secondary thiol was employed in a range of reactions with a commercially available epoxide. This particular route was only examined for PS. The second route, Route 2 Scheme 1, was a one-pot procedure involving hydride cleavage of the TCT end-groups in the presence of Me2PPh and epoxide.
Clearly, the success of Route 1 relies on the ability to isolate the thiol-terminated (co)polymer. The isolation of ‘pristine’ PS–SH can be conveniently confirmed using a combination of UV-Vis spectrophotometry and SEC, Fig. 2. Prior to hydrazinolysis PS–S(CS)Ph exhibits an intense absorption band at ca. 300 nm that is attributed to the n–π* transition associated with the CS bond, Fig. 2 (black line). Upon cleavage this functionality is removed from the chain end and this band disappears (blue line). While UV-Vis confirms the successful conversion of the TCT group to the free thiol SEC is employed to confirm the absence of any disulfide arising from the oxidation of the newly formed macromolecular thiols. Fig. 2, inset, shows the SEC traces (normalized RI response) for the precursor PS–S(CS)Ph and the resulting PS–SH homopolymer. Importantly, after hydrazinolysis a small shift to lower molecular weight is observed and in both instances the distributions are unimodal indicating the absence of any coupled species.
 | ||
| Fig. 2
UV-Vis spectra of the parent PS homopolymer before and after treatment with hydrazine highlighting the n–π* transition associated with the CS bond and SEC traces (inset, normalized RI response) of the same homopolymer before and after dithioester end-group cleavage. | ||
With PS–SH successfully isolated and characterized its reaction with 2-(but-3-en-1-yl)oxirane, EP1Fig. 3, was initially examined. The presence of a CC bond in EP1 was considered beneficial for two reasons. Firstly, it was anticipated that the CC bond would exhibit orthogonal reactivity under the conditions employed for macromolecular thiolysis thus presenting the opportunity for conducting sequential reactions (thiol–oxirane followed by thiol–ene) and secondly, thiolysis of EP1 would yield an end-modified polymer with an unreacted ene bond thus facilitating convenient end-group analysis by 1H NMR spectroscopy.
 | ||
| Fig. 3 Chemical structures of commercially available epoxides employed in this study. | ||
PS–SH was reacted with EP1 in the presence of two different catalysts—1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as an organobase catalyst and ZnCl2 as a Lewis acid catalyst. Consider first reactions performed in the presence of ZnCl2. Chain-end modifications were performed with different loadings of ZnCl2 (10 or 15 mol%) for varying lengths of time and at two different temperatures, Table 1. In all instances thiolysis of EP1 was successful although quantitative chain end functionalization was not achieved as evidenced by 1H NMR spectroscopy. The highest degree of modification (88%) was achieved at 60 °C with 15 mol% ZnCl2 (based on epoxide) for 96 h. Extending the reaction time further appeared to have no effect on the reaction yield.
| Sample | Catalyst (mol%) | Reaction time | Reaction temp/°C | M n a | M w/Mna | Conversion (%)b |
|---|---|---|---|---|---|---|
| a As determined by SEC. b As determined by 1H NMR spectroscopy. | ||||||
|
PS–S(CS)Ph |
— | — | — | 3540 | 1.08 | — |
| PS–SH | — | — | — | 3560 | 1.09 | — |
| 1 | ZnCl2, 10 | 48 | 60 | 3740 | 1.07 | 82 |
| 2 | ZnCl2, 15 | 72 | RT | 3700 | 1.08 | 74 |
| 3 | ZnCl2, 15 | 96 | 60 | 3680 | 1.08 | 88 |
| 4 | ZnCl2, 15 | 120 | 60 | 4370 | 1.05 | 85 |
| 5 | DBU | 48 | 60 | 3790 | 1.12 | 74 |
| 6 | DBU | 48 | 60 | 3620 | 1.15 | 89 |
As a representative example, Fig. 4 shows the 1H NMR spectrum of the product obtained from the reaction of PS–SH and EP1.
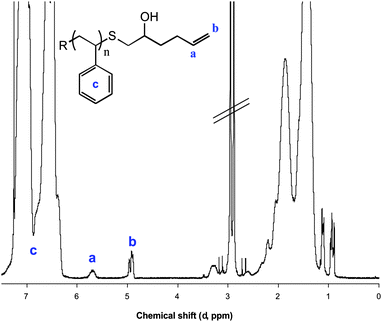 | ||
| Fig. 4 1H NMR spectrum, recorded in CDCl3, of PS–SH after reaction with EP1 highlighting the vinyl hydrogens of the ene end-group. | ||
The key features confirming end-group functionalization are the signals labelled a and b and are assigned to the vinylic hydrogens of the ene bond originally associated with EP1. A ratio of the integral of either a or b with c, the aromatic hydrogens of the main polymer chain, yielded, in this instance, a degree of modification of 88%.
Importantly, the molecular weight distributions of the resulting end-functionalized PS are unaffected by the thiolysis reaction with all distributions remaining unimodal and narrow, Table 1 and Fig. 5, with, in all instances, an increase in measured molecular weight (Mn) being observed after the thiolysis reaction. The experimentally determined SEC traces (normalized RI response) for PS–S(CS)Ph, PS–SH and examples of the product obtained from the reaction of PS–SH with EP1 are shown in Fig. 5.
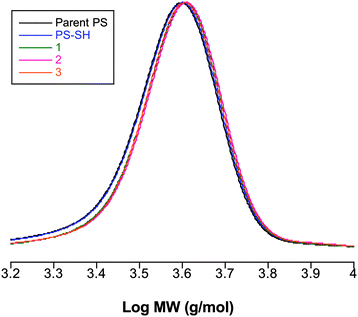 | ||
| Fig. 5
SEC traces (normalized RI responses) for PS–S(CS)Ph, PS–SH and three examples of products obtained from the reaction of PS–SH with EP1 in the presence of ZnCl2. | ||
While the reaction of PS–SH with EP1 in the presence of ZnCl2 was clearly successful yielding the desired ene–OH terminal polymeric species, although not in a click fashion, the use of a Lewis acid metal catalyst and less than quantitative conversion prompted us to examine alternative conditions for effecting chain-end modification. Specifically, the reaction of PS–SH with EP1 in the presence of DBU and Me2PPh was briefly examined, Table 1 entries 5 and 6.
While chain-end functionalization was successful, although not quantitative, as judged by 1H NMR spectroscopy, in both instances there was clear evidence of coupled species in the SEC traces, Fig. 6, suggesting that macromolecular thiolysis with DBU is an even less satisfactory option than the use of ZnCl2. Given these observations we decided to explore Route 2—a one-pot cleavage/thiolysis protocol.
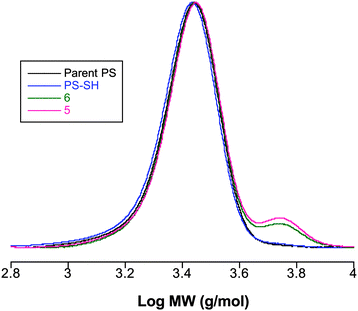 | ||
| Fig. 6
SEC traces (normalized RI responses) for PS–S(CS)Ph, PS–SH and two examples of products obtained from the reaction of PS–SH with EP1 in the presence of DBU. | ||
Initially the one-pot reaction of PDEAM–S(CS)Ph with seven small molecule epoxides, namely EP1, 2-butyloxirane (EP2), 2-vinyloxirane (EP3), 2,2-dimethyloxirane (EP4), 3-vinyl-7-oxabicyclo[4.1.0]heptane (EP5), 1-methyl-3-(prop-1-en-2-yl)-7-oxabicyclo[4.1.0]heptane (EP6) and 2-((4-nonylphenoxy)-methyl)oxirane (EP7) was examined. In all instances reactions were conducted at RT for either 2, 3 or 4 days, Table 2.
| Sample | Epoxide | Reaction time | Reaction temp/°C) | M n a | M w/Mna | Conversion (%)b |
|---|---|---|---|---|---|---|
| a As determined by SEC. b As determined by 1H NMR spectroscopy. | ||||||
|
PDEAM–S(CS)Ph |
— | — | — | 1790 | 1.06 | — |
| 7 | EP1 | 48 | RT | 1730 | 1.08 | >98 |
| 8 | EP2 | 72 | RT | 2240 | 1.07 | — |
| 9 | EP3 | 96 | RT | 2400 | 1.10 | >99 |
| 10 | EP4 | 72 | RT | 2360 | 1.10 | — |
| 11 | EP5 | 48 | RT | 1730 | 1.09 | >99 |
| 12 | EP6 | 72 | RT | 2500 | 1.07 | >99 |
| 13 | EP7 | 48 | RT | 1960 | 1.07 | >91 |
As with EP1, EP3, EP5, EP6 and EP7 contain functionality that was anticipated to facilitate end-group analysis by 1H NMR spectroscopy. With the exception of EP7, NMR analysis indicated essentially quantitative reaction with calculated conversions generally >99%. However, the measured conversion for the reaction of PDEAM–S(CS)Ph with EP7 of >91% was still higher than any observed in the reactions of PS–SH with EP1via Route 1, implying that Route 2 is the preferred method for effecting macromolecular thiolysis.
As a representative example, Fig. 7, shows the 1H NMR spectrum, recorded in CDCl3, obtained for the product derived from the reaction of PDEAM–S(CS)Ph with EP6. A ratio of the signals labeled a and b, assigned to the vinyl hydrogens of the end-group and the methylene hydrogens attached to N in the DEAM repeat unit respectively, gives a calculated degree of functionalization of >99%. Fig. 8 shows the experimentally determined SEC traces for the products obtained from the reaction between PDEAM–S(CS)Ph and EP1, EP5 and EP7. All traces are narrow and unimodal. Successful end-group modification was also confirmed qualitatively viaFTIR spectroscopy, see ESI†.
 | ||
| Fig. 7
1H NMR spectrum, recorded in CDCl3, of the product obtained from the reaction of PDEAM–S(CS)Ph and EP6 highlighting the ene end-group. | ||
Many water-soluble (co)polymers exhibit inverse temperature-dependent water-solubility, i.e. a lower critical solution temperature (LCST) or cloud-point. LCSTs can range from just above 0 °C to just below 100 °C and depend on factors such as the chemical structure of the polymer, the molecular weight and for low molecular weight (co)polymers the nature of the end-groups.60–62 One of the most widely studied temperature responsive polymers is poly(N-isopropylacrylamide) (PNIPAM) in part due to the fact that its LCST is close to physiological temperature. A convenient alternative to PNIPAM, with an essentially identical LCST, is PDEAM.63–65 Given the low molecular weight of the PDEAM homopolymer used in these end-group functionalization studies it was anticipated that a discernible effect on the LCST would be observed, in a manner similar to that recently reported for end-group
 | ||
| Fig. 8 SEC traces (normalized RI response) for PDEAM–S(CS)Ph and the products obtained from reaction with EP1, EP5 and EP7. | ||
Fig. 9 shows the measured cloud-point curves for PDEAM–S(CS)Ph as well as the end-functional products (PDEAM–EPX) obtained from reactions with EP1–EP7 performed in the presence of NaBH4. Reaction of the thiol/thiolate end-groups with a given epoxide results in the introduction of both hydrophilic (hydroxy) and hydrophobic (alkyl or aryl) functionality at the ω-terminus. Generally, the effect on the measured cloud-points is consistent with the hydrophobic contribution assuming the relative hydrophilic contribution from the OH groups is approximately the same. The parent PDEAM–S(CS)Ph has a measured LCST of ∼34 °C—slightly higher than a previously reported value for a PDEAM homopolymer with a dithioester end-group49 although there is a significant difference in the molecular weights which likely accounts for the difference. Of the seven epoxides used EP7 is the most hydrophobic and the product obtained from its reaction with PDEAM–S(CS)Ph (PDEAM–EP7) exhibits the lowest LCST of ∼28 °C. All other products gave measured LCSTs that were higher than the parent homopolymer indicating an increase in overall hydrophilicity. The products obtained from EP1, EP2, EP5 and EP6 all exhibited approximately the same LCST of ∼41 °C. The highest LCSTs were observed for the lowest molecular weight epoxides namely EP3 and EP4 with measured values of ∼48 and 46 °C respectively.
 | ||
| Fig. 9
Experimentally determined cloud-point curves for the parent PDEAM homopolymer and the products obtained from the reaction of PDEAM–S(CS)Ph with EP1–EP7. | ||
Given the success of Route 2 as a means of functionalizing PDEAM the same conditions were evaluated for the end-group modification of PS–S–(CS)Ph. Reactions were performed for 3 days at both RT and 60 °C with EP1. While degrees of functionalization approaching 80% were achieved in all instances there was evidence of coupled products as determined by SEC, Fig. 10.
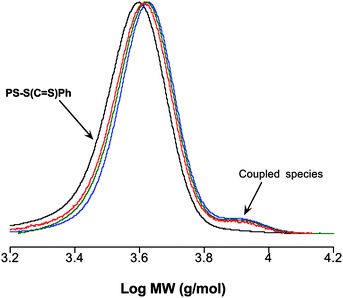 | ||
| Fig. 10
SEC traces (normalized RI responses) for PS–S(CS)Ph and the products obtained from the reaction with EP1 in the presence of NaBH4, showing the presence of coupled species. | ||
In addition to the small molecule monofunctional epoxide reactions with di- and tri-functional epoxides were also examined briefly. Resorcinol diglycidylether (EP8) and bisphenol A diglycidylether (EP9) were chosen as difunctional epoxides with the intent of preparing end-functional polymers with an intact epoxy functionality. Castor oil (EP10), tris(2,3-epoxypropyl)isocyanurate (EP11) and trimethylolpropane triglycidylether (EP12) were chosen as trifunctional oxiranes with the intent of examining the possibility of preparing 3-arm star polymers. In all instances PDEAM–S(CS)Ph was employed with NaBH4 as the cleavage agent in the presence of an appropriate oxirane. Generally, modifications appeared to proceed smoothly, as judged by SEC, although in all instances end-group analysis by NMR spectroscopy proved problematic primarily due to the lack of an appropriate signal for effective integration.
Fig. 11A shows the measured SEC traces for the PDEAM–S(CS)Ph homopolymer along with the product obtained from the reaction with an excess of EP8, resorcinol diglycidylether. After reaction a shift to higher molecular weight is observed while the molecular weight distribution remains unimodal and narrow. Importantly there is no evidence of coupled products that could arise from either thiolate–thiolate oxidation leading to disulfide formation or from reaction at both epoxy functional groups on a single EP8 molecule. Reaction of PDEAM–S(CS)Ph with EP12, trimethylolpropane triglycidylether, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 functional group ratio in the presence of NaBH4 yields a product whose SEC trace is shown in Fig. 11B. A pronounced shift to higher molecular weight is observed, consistent with the formation of the target star products, although the resulting molecular weight distribution is bimodal, perhaps indicating a mixture of 2- and 3-arm products.
:1 functional group ratio in the presence of NaBH4 yields a product whose SEC trace is shown in Fig. 11B. A pronounced shift to higher molecular weight is observed, consistent with the formation of the target star products, although the resulting molecular weight distribution is bimodal, perhaps indicating a mixture of 2- and 3-arm products.
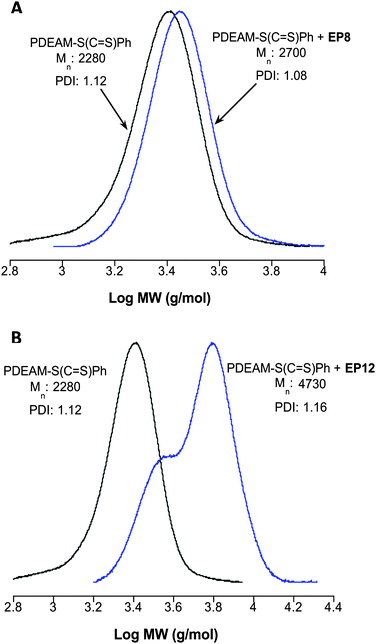 | ||
| Fig. 11
SEC traces (normalized RI signals) for PDEAM–S(CS)Ph and the product obtained from reaction with EP8 (A) and PDEAM–S(CS)Ph and star products obtained from reaction with EP12 (B). | ||
Summary/conclusions
Herein we have described our preliminary observations regarding the ω-end group functionalization of polystyrene and poly(N,N-diethylacrylamide)via the macromolecular thiolysis of mono-, di- and tri-functional oxiranes. Hydrazinolysis of PS–S(CS)Ph to yield PS–SH followed by reaction with EP1 in the presence of ZnCl2 gave the target products in yields up to 88% while maintaining a narrow, unimodal molecular weight distribution. In contrast, catalysis with DBU, while also giving high yields of the target end functional homopolymer, also yielded a significant fraction of coupled species as determined by SEC. Similar observations were made for PS–S(CS)Ph when a one-pot protocol was employed involving end-group cleavage by NaBH4 in the presence of EP1. The one-pot reaction of PDEAM–S(CS)Ph with a series of monofunctional oxiranes in the presence of NaBH4 yielded the target materials in essentially quantitative yield highlighting the difference in reactivity between macromolecular thiols derived from PS and PDEAM. The effect of the newly introduced end-groups on the lower critical solution temperature for the PDEAM materials was examined with the cloud point being tunable over the range ∼27 to 46 °C. Reaction of PDEAM–S(CS)Ph with difunctional epoxides in the presence of NaBH4 appears to yield the target materials in high-to-quantitative yield although end-group analysis was problematic. Treatment of PDEAM–S(CS)Ph with a trifunctional epoxide in a 1:1 functional group ratio appears to yield a mixture of 2- and 3-arm star polymer as judged by SEC. While such macromolecular thiolysis reactions can give quantitative formation of desired products these appear not to be as facile as other macromolecular thiol-based reactions and care should be employed when adopting such chemistries.
References
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, Aust. J. Chem., 2006, 59, 661–662 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- E. Rizzardo, M. Chen, B. Chong, G. Moad, M. Skidmore and S. H. Thang, Macromol. Symp., 2007, 248, 104–116 CrossRef CAS.
- G. Moad and S. H. Thang, Aust. J. Chem., 2009, 62, 1379–1381 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- J. Xu, J. He, D. Fan, X. Wang and Y. Yang, Macromolecules, 2006, 39, 8616–8624 CrossRef CAS.
- W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. Cao and Z. An, Macromol. Rapid Commun., 2010, 31, 1444–1448 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer International, 2011, 60, 9–25 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- B. Le Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- A. J. D. Magenau, T. R. Hartlage and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5505–5513 CrossRef CAS.
- M. Li, P. De, H. M. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- K. Y. van Berkel, A. M. Piekarski, P. H. Kierstead, E. D. Pressly, P. C. Ray and C. J. Hawker, Macromolecules, 2009, 42, 1425–1427 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- S. S. Naik, J. W. Chan, C. Comer, C. E. Hoyle and D. A. Savin, Polym. Chem., 2011, 2, 303–305 RSC.
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937–4942 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS.
- G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291–6293 RSC.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- J. D. Flores, J. Shin, C. E. Hoyle and C. L. McCormick, Polym. Chem., 2010, 1, 213–220 RSC.
- R. M. Hensarling, S. B. Rahane, A. P. LeBlanc, B. J. Sparks, E. M. White, J. Locklin and D. L. Patton, Polym. Chem., 2011, 2, 88–90 RSC.
- J. T. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2010, 43, 20–24 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3940–3948 CrossRef CAS.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- V. Grazu, O. Abian, C. Mateo, F. Batista-Viera, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2003, 4, 1495–1501 CrossRef CAS.
- M. S. Abaee, M. M. Mojtahedi, H. Abbasi and E. R. Fatemi, Synth. Commun., 2008, 38, 282–289 CrossRef.
- D. Amantini, F. Fringuelli, F. Pizzo, S. Tortoioli and L. Vaccaro, Synlett, 2003, 2292–2296 CAS.
- J. Zhu, R. Li, Z. Ge, T. Cheng and R. Li, Chin. J. Chem., 2009, 27, 791–796 CrossRef CAS.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
- X. Jiang and B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3707–3721 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- Y. Katsumoto, Y. Etoh and N. Shimoda, Macromolecules, 2010, 43, 3120–3121 CrossRef CAS.
- M. Panayiotou, C. Pohner, C. Vandevyver, C. Wandrey, F. Hilbrig and R. Freitag, React. Funct. Polym., 2007, 67, 807–819 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information See DOI: 10.1039/c1py00046b |
| This journal is © The Royal Society of Chemistry 2011 |