Photochemical stability and photovoltaic performance of low-band gap polymers based on dithiophene with different bridging atoms†
Martin
Helgesen
*a,
Thomas J.
Sørensen
b,
Matthieu
Manceau
a and
Frederik C.
Krebs
a
aRisø National Laboratory for Sustainable Energy, Technical University of Denmark, Frederiksborgvej 399, DK-4000, Roskilde, Denmark. E-mail: manp@risoe.dtu.dk
bNano-Science Center and Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100, København Ø, Denmark
First published on 28th March 2011
Abstract
New low-band gap polymers based on dithienylbenzothiadiazole (DBT) and dithiophene with different bridging atoms have been synthesized and explored in a comparative study on the photochemical stability and photovoltaic performance. Two differently modified DBT units were exploited, namely 5,6-bis(tetradecyloxy)-4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (DBT1) and 4,7-bis(4-dodecylthiophen-2-yl)benzo[c][1,2,5]thiadiazole (DBT2). In thin films the polymers had optical band gaps in the range of 1.51–1.70 eV where the DBT2 based polymers are red shifted 61–81 nm compared to the DBT1 based polymers indicating greater interchain packing when the side chains are situated on the thienyl groups compared to on the benzothiadiazole unit. The best photovoltaic devices based on blends of polymer and [6,6]-phenyl C61butyric acid methyl ester (PCBM) were prepared with polymers based on the DBT1 unit giving efficiencies up to 2.3%. The photochemical stability was measured by the amount of absorbed photons under 1 sun versus the ageing time for each polymer, which clearly shows that the two polymers containing a 4,4-bis(2-ethylhexyl)-4H-cyclopenta[1,2-b:5,4-b′]dithiophene (CPDT) unit are by far the most unstable. Substitution of the bridging carbon atom with silicon results in a significant stability improvement by a factor 5.
Introduction
Polymer solar cells (PSCs)1–5 are viewed as a very promising candidate for low cost photovoltaics because they enable preparation under ambient conditions using low temperature roll-to-roll coating and printing6–13 of liquid polymer solutions onto flexible substrates. These techniques enable fast deposition of polymers onto flexible substrates over large areas which can realize the presumed very low production cost of PSCs compared to the more expensive inorganic solar cells. Research in the field of PSCs during the past decade has been intensively focused on improving the device power conversion efficiency that now exceeds 8% for small area devices.14 Highly efficient polymers are certainly significant for practical use in large-scale PSC production but will only be attractive candidates if they also provide a good processability, a sufficient photochemical stability and device stability.6,15In terms of materials for the photoactive layer, low-band gap conjugated polymers16,17 blended with a soluble fullerene derivative promise efficient light harvesting across a broad area of the solar spectrum. Low-band gap donor–acceptor polymers based on 2,1,3-benzothiadiazole and dithiophene-based units have been extensively studied over the years. Namely poly[2,6-(4,4-dialkyl-4H-cyclopenta-[2,1-b;3,4-b′]-dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (PCPDTBT) blended with [6,6]-phenyl-C71-butyric acid methyl ester ([70]PCBM) has showed high efficiencies up to 5.5% when a processing additive (octanedithiol) was used to alter the nanomorphology and increase the fill factor (FF) beyond 50%. Despite the promising performance, there are some concerns about the stability of this polymer system due to the presence of the quaternary carbon site in the five-membered ring that could readily undergo oxidation as reported for polyfluorenes.18,19 In order to improve the stability one could explore new derivatives like the silole (silacyclopentadiene) moiety that has recently been investigated in novel conjugated systems20–24 showing altered properties with respect to their carbon analogues. The silole analogue of PCPDTBT has been tested in bulk heterojunction solar cells showing higher carrier mobility20 and enhanced interchain packing25,26 which has resulted in power conversion efficiencies up to 5.9%.27 Enhanced carrier mobility has also been reported when the carbon atoms on the 9-position of the fluorene units of poly[(9,9-dialkylfluorene)-2,7-diyl-alt-(4,7-bis(2-thienyl)-2,1,3-benzothiadiazole)-5,5′-diyl] (PFDTBT) were substituted with silicon atoms which resulted in a significant improvement of the photovoltaic properties of the material.23
To further explore silicon based polymers with respect to their carbon analogues, we present a comparative study on the photochemical stability and photovoltaic performance of films comprising four new low-band gap polymers based on 4,7-di-2-thienyl-2,1,3-benzothiadiazole28 (DBT) and dithiophene with different bridging atoms (Scheme 1). Two differently modified DBT units were exploited bearing solubilising chains on either the thienyl groups (DBT2) or on benzothiadiazole (DBT1) which has a pronounced effect on the molecular weight, optical properties and photovoltaic performance in polymer/PCBM solar cells. In terms of photochemical stability the dithiophene unit has the major influence on how fast the polymers are ageing under illumination which is investigated together with the optical, electrical and photovoltaic properties.
 | ||
| Scheme 1 Synthesis of the polymers PCPDT-DBT1, PCPDT-DBT2, PSDT-DBT1 and PSDT-DBT2. | ||
Results and discussion
The polymers were prepared via Stille cross-coupling using the catalyst system Pd2dba3/P(o-tolyl)3. Copolymerisation of 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(tetradecyloxy)benzo[c][1,2,5]thiadiazole29 and 4,7-bis(5-bromo-4-dodecylthiophen-2-yl)benzo[c][1,2,5]thia-diazole30 with (4,4-bis(2-ethylhexyl)-4H-cyclopenta[1,2-b:5,4-b′]dithiophene-2,6-diyl)bis(trimethylstannane)31 or 4,4-dihexyl-2,6-bis(trimethylstannyl)-4H-silolo[3,2-b:4,5-b′]dithiophene22 gives the polymers PCPDT-DBT1, PCPDT-DBT2, PSDT-DBT1 and PSDT-DBT2 in high yield (82–97%) as dark blue solids. Generally a higher molecular weight was reached when coupling with the DBT1 unit compared to DBT2 (Table 1) which is ascribed to increased solubility of the polymer during polymerization. At room temperature the polymers based on the CPDT unit (PCPDT-DBT1 and PCPDT-DBT2) demonstrate the best solubility in organic solvents such as chloroform and chlorobenzene due to enhanced solubility of the ethylhexyl side chains whereas PSDT-DBT1 and PSDT-DBT2 show good solubility only in hot chlorinated solvents. When comparing to the earlier reported polymer poly-[2,6-(4,4-bis(2-ethylhexyl)-4H-cyclopenta-[2,1-b:3,4-b′]dithiophene-alt-4,7- bis(thiophen-2-yl)benzo-2,1,3-thiadiazole] (PCPDTTBTT)28 that has the same backbone but does not carry solubilising chains on the DBT unit the molecular weight is enhanced significantly for the polymers reported in this work.| Polymer | M n/g mol−1 | PDI | α(λmax)/L g−1 cm−1 | Solution | Film | ||||
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm | λ onset/nm | E g/eV | λ max/nm | λ onset/nm | E g/eV | ||||
| PCPDT-DBT1 | 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
2.2 | 30 | 600 | 683 | 1.82 | 610 | 729 | 1.70 |
| PCPDT-DBT2 | 17600 |
1.9 | 27 | 596 | 712 | 1.74 | 633 | 790 | 1.57 |
| PSDT-DBT1 | 26400 |
2.8 | 37 | 597 | 700 | 1.77 | 615 | 738 | 1.68 |
| PSDT-DBT2 | 22400 |
2.4 | 27 | 590 | 723 | 1.71 | 656 | 819 | 1.51 |
The absorption spectra of the polymers in the chloroform solution and in the thin film are shown in Fig. 1. In solution the optical band gaps, defined by the onset of absorption, of the polymers are rather similar ranging from 1.71–1.82 eV (Table 1) but a general observation is that a lower band gap is reached when the side chains are shifted from benzothiadiazole to the thienyl groups since the electron donating alkoxy chains on benzothiadiazole will reduce the acceptor strength. Though, the polymers with the side chains situated on the benzothiadiazole exhibit the highest extinction coefficient (Table 1). The effect of the side chain position and its chemical nature is more pronounced in the solid state where the onset of absorption is red shifted 61–81 nm for PCPDT-DBT2 and PSDT-DBT2 in contrast to PCPDT-DBT1 and PSDT-DBT1 which can be explained by a greater tendency to π-stack in the solid state. Also the choice of the bridging atom in the dithiophene unit has a minor effect on the band gap which is reduced when carbon is substituted with silicon indicating a higher degree of π-delocalization in the silicon bridged polymers. Though, it should be noted that the silolodithiophene unit bears less bulky hexyl chains compared to the ethylhexyl chains on the CPDT unit. PCPDT-DBT1 and PSDT-DBT1 show vibronic fine structure at 625 nm in the solid state and a weak vibronic transition may also account for the observed shoulder of PSDT-DBT2 at around 700 nm (Fig. 1b).
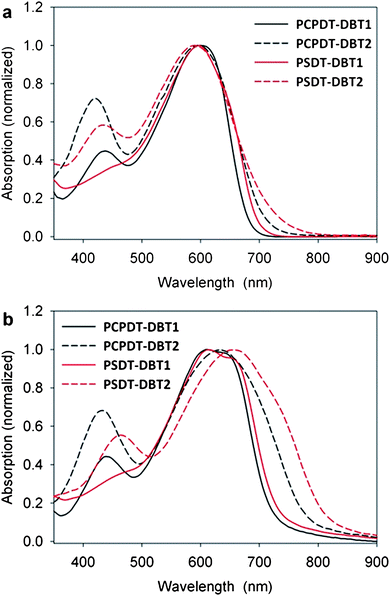 | ||
| Fig. 1 (a) UV-vis absorption spectra of the polymers in chloroform solution and (b) in thin film. | ||
To obtain further information about the electronic bands the electrochemical properties of the polymers were investigated by using cyclic voltammetry (CV). The HOMO/LUMO energy levels deduced from their voltammograms are shown in Fig. 2. The HOMO energy levels of the polymers lie relatively close but PCPDT-DBT1 and PCPDT-DBT2 are found at lower levels compared to the silicon bridged polymers which are beneficial for reaching a higher open-circuit voltage in photovoltaic devices.
 | ||
| Fig. 2 HOMO and LUMO energy levels of the polymers and PCBM. | ||
The oxidation of the polymer films occurs in two steps. The voltammogram of PCPDT-DBT1 that has the best behaved electrochemistry shows that the first oxidation gives a resolved peak while the shape of the second peak is broader (Fig. 3). After the second peak the film is dissolved which is observed in the voltammogram as the sharp feature at 1.10 V. Variation of the scan rate shows that the oxidation is well behaved and the current scales linearly with the scan rate as expected32 (Fig. 4a). Furthermore, Fig. 4a shows that the peak position shifts as the scan rate is increased which indicates that the electron transfer to the polymer happens with a rate comparable to the scan rate. For PCPDT-DBT1 and PCPDT-DBT2 the peaks corresponding to the first oxidations are narrower than what is observed for the following scans where the change of the onset potential is 10–50 mV to a lower potential. The oxidation of PSDT-DBT1 and PSDT-DBT2 are fully irreversible. The films simply dissolve as soon as oxidation occurs. Fig. 4b shows voltammograms of PSDT-DBT2 at different scan rates and the desorption of the film is clearly seen when the voltammograms are normalized with the scan rates. Only one oxidation peak is observed at slow scan rates while a second low potential peak becomes visible when the scan rate is increased.
 | ||
| Fig. 3 Cyclic voltammograms of polymer films on ITO in 0.1 M Bu4NPF6/MeCN at a scan rate of 100 mV s−1. | ||
 | ||
| Fig. 4 (a) Cyclic voltammograms of PCPDT-DBT1 and (b) PSDT-DBT2 films on ITO in 0.1 M Bu4NPF6/MeCN with varying scan rates. | ||
The photovoltaic performance of the polymers was tested in bulk heterojunction polymer/PCBM solar cells with the conventional device architecture ITO/PEDOT:PSS/polymer:PCBM/Al. The optimized solar cell efficiencies are summarized in Table 2 and the obtained current–voltage curves of the polymer:PCBM solar cells are presented in Fig. 5a. As was expected from the HOMO energy levels (Fig. 2) PCPDT-DBT1 and PCPDT-DBT2 have the highest Voc of 0.68 V.
| Weight ratio | Annealing | V oc/V | J sc/mA cm−2 | FF | η/% |
|---|---|---|---|---|---|
|
PCPDT-DBT1:PCBM = 1:2 |
— | 0.68 | 7.42 | 0.45 | 2.27 |
|
PCPDT-DBT2:PCBM = 1:3 |
— | 0.68 | 6.07 | 0.42 | 1.73 |
|
PSDT-DBT1:PCBM = 1:3 |
100 °C (2 min) | 0.65 | 6.39 | 0.48 | 1.99 |
|
PSDT-DBT2:PCBM = 1:1 |
100 °C (2 min) | 0.59 | 5.79 | 0.37 | 1.26 |
 | ||
| Fig. 5 (a) J–V characteristics of polymer:PCBM solar cells measured under 100 mW cm−2 white light (b) IPCE of polymer:PCBM solar cells. | ||
Generally the best devices were prepared with polymers based on the DBT1 unit which was mainly due to a higher current density (Jsc) and fill factor (FF). PCPDT-DBT1:PCBM solar cells had moderate fill factors of 0.45 and current densities of 7.42 mA cm−2 that resulted in the highest power conversion efficiencies of up to 2.3% which is very close to earlier reported efficiencies of the polymer PCPDTTBTT (η = 2.1%) with the same backbone reported by Moulé et al.28 The somewhat higher performance of the polymers based on the DBT1 unit is also reflected in the incident photon to current efficiency (IPCE) which reaches a maximum IPCE of 41% for PCPDT-DBT1 with a photoresponse up to 750 nm (Fig. 5b). The DBT2 based polymers have a somewhat broader photoresponse compared to PCPDT-DBT1 and PSDT-DBT1 in agreement with the absorption spectra (Fig. 1b) but the IPCE is up to 19% lower. Finally it should be noted that the silicon bridged polymer:PCBM devices require annealing to reach their maximum performance.
As mentioned above stable photovoltaic application is also an important aspect that novel polymer materials have to possess in order to be considered as attractive candidates for large-scale PSC production. In order to quickly evaluate the long-term durability of the newly synthesized polymers their photochemical stability under 1 sun was measured. Fig. 6 presents the evolution of the normalized amount of absorbed photonsversusageing time for each polymer. From these results, it is obvious that the two polymers containing the CPDT unit (PCPDT-DBT1 and PCPDT-DBT2) are by far the most unstable. This low photochemical stability is due to the presence of the quaternary carbon site that can be readily oxidized.33,34 Substitution of the carbon atom with silicon results in a significant stability improvement and PSDT-DBT2 (respectively PSDT-DBT1) proved to be about 5 times more stable than PCPDT-DBT2 (respectively PCPDT-DBT1) under the same conditions. HOMO levels of the silicon bridged polymers were estimated to be slightly higher than the carbon bridged ones (Fig. 2), meaning that the stability enhancement does not result from a deeper HOMO level of the silicon bridged derivatives.35 It could however be explained by the reduced oxidizability of the silicon site compared to the carbon one. One can also notice that the replacement of the ether side chains by alkyl ones has no significant influence on the photochemical stability of the carbon bridged derivatives. Once again this can be explained by the presence of the CPDT unit which completely limits sample durability. Conversely, a slight stability increase is recorded for the silicon bridged compounds. In this case, the backbone stability is much higher and the side chain effect is not negligible. It is however quite surprising that substitution of ethers by alkyl groups does not lead to a more important increase in stability as ether side chains are known to be rather unstable under irradiation. Though, it should be noted here that the comparison between the samples is not totally straightforward as the side chains are located on different units. Alkyl groups are on a thiophene moiety (i.e. an electron donor) while ether side chains are on a benzothiadiazole unit (i.e. an electron acceptor) and the positioning of a given side chain either on a donor or on acceptor certainly affects the stability. For instance, this modifies the bond strength between the backbone and the substituent and hence the cleavability of this bond. To the best of our knowledge this point has not been investigated yet and its importance is then difficult to estimate before further studies are made.
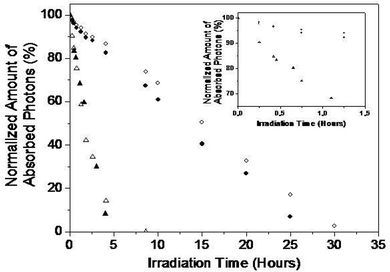 | ||
| Fig. 6 Evolution of the normalized amount of absorbed photons during photochemical ageing of (▲) PCPDT-DBT1, (△) PCPDT-DBT2, (●) PSDT-DBT1 and (○) PSDT-DBT2 in air (AM 1.5G, 1000 W m−2, 85 °C). | ||
Conclusion
Four new low-band gap polymers based on 4,7-di-2-thienyl-2,1,3-benzothiadiazole (DBT) and dithiophene with different bridging atoms (carbon and silicon) have been synthesized and tested in polymer:PCBM solar cells. Two different DBT units were exploited bearing solubilising chains on either the thienyl groups (DBT2) or on benzothiadiazole (DBT1). A lower band gap is reached with the DBT2 based polymers where the onset of absorption in thin films is red shifted 61–81 nm for PCPDT-DBT2 and PSDT-DBT2 in contrast to PCPDT-DBT1 and PSDT-DBT1 (Table 1). The choice of bridging atom in the dithiophene unit also has a minor effect on the band gap which is reduced when carbon is substituted with silicon indicating a higher degree of π-delocalization in the silicon bridged polymers. In solar cells PCPDT-DBT1 and PCPDT-DBT2 have the highest Voc of 0.68 V as was expected since they exhibit the lowest HOMO levels as measured with CV. Generally the best photovoltaic devices were prepared with polymers based on the DBT1 unit giving efficiencies up to 2.3%. Finally, measuring the amount of absorbed photons under 1 sun versus the ageing time for each polymer clearly shows that the two polymers containing a CPDT unit are by far the most unstable due to the presence of the quaternary carbon site that can be readily oxidized. Substitution of the bridging carbon atom with silicon results in a significant stability improvement by a factor 5 making the silicon bridged polymers the best choice for stable photovoltaic application.Experimental section
General procedure for the Stille polymerization
:10). Finally the polymer was filtered and dried in vacuum at 50 °C for 24 hours. Yield: 331 mg (86%), dark blue solid. 1H NMR (250 MHz, CDCl3) δ = 8.63–8.48 (br, 2H), 7.33–7.28 (br, 2H), 7.23–7.13 (br, 2H), 4.45–3.97 (br, 4H), 2.17–1.83 (br, 8H), 1.62–1.18 (m, 46H), 1.15–0.93 (m, 16H), 0.92–0.82 (m, 6H), 0.82–0.62 (m, 12H). SEC (CHCl3): Mn = 31000 g mol−1, PDI = 2.2.
600 g mol−1, PDI = 1.9.
400 g mol−1, PDI = 2.8.
400 g mol−1, PDI = 2.4.
Polymer solar cell fabrication and analysis
Photovoltaic devices were made by spin coating PEDOT:PSS (Aldrich, 1.3 wt% aqueous solution) onto precleaned, patterned indium–tin oxide (ITO) substrates (9–15 Ω per square) (LumTec) followed by annealing at 140 °C for 5 min. The active layer was deposited by spin coating a blend of the polymer and PCBM dissolved in chlorobenzene (25 mg ml−1). Finally the counter electrode of aluminium was deposited by vacuum evaporation at 2–3 × 10−6 mbar. The active area of the cells was 0.5 cm2. A relatively large active area is used since it has been reported that power conversion efficiencies obtained on areas smaller than around 0.3 cm2 may become very size-dependent.36,37I–V characteristics were measured under AM1.5G corresponding to 100 mW cm−2 white light from a multi-wavelength high-power LED array using a Keithley 2400 source meter. IPCE spectra were recorded on the same solar test platform38 with the LED based illumination system.Electrochemistry
Cyclic voltammetry was carried out in a conventional three electrode setup, with the polymer film on ITO acting as a working electrode. The polymer films were spin coated from 10 mg ml−1chlorobenzene solution at 1000–1500 rpm onto patterned indium–tin oxide (ITO) substrates (9–15 Ω per square) (LumTec) prior to measurements. Care was taken to make sure that the conductive ITO surface with polymer was facing the reference and counter electrode. A platinum wire counter electrode was used along a silver wire reference electrode. The potential of the reference electrode was referenced to the ferrocene/ferrocenium and the decamethylferrocene/decamethylferrocenium redox couples. The voltammograms were recorded using a 0.1 M tetrabutylammonium hexafluorophosphate in acetonitrile electrolyte. The onset potential is determined as the intersect of the baseline and a tangent to the first peak. Only values from the first sweep on a film were used as the film is changed or destroyed by the first oxidation. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of polymers were calculated from the onset oxidation potential (Eox) according to the equations HOMO = −(Eox + 4.8)(eV) and LUMO = HOMO + Egopt (eV).Photochemistry
Pure polymer samples—PCPDT-DBT1, PCPDT-DBT2, PSDT-DBT1 and PSDT-DBT2—were spin-coated in air on glass slides from chlorobenzene solutions. The solution concentrations and spinning speeds were adjusted to get a maximum peak absorbance of about 0.25 for each material. The low absorbance was chosen to quickly compare the materials photochemical stability. Samples were aged under 1 sun in air using a standard solar simulator from Steuernagel Lichttechnik (KHS 575, AM 1.5G, 1000 W m−2, 85 °C). To monitor the degradation progress, samples were removed periodically and UV-visible spectra were recorded from 200 to 1100 nm using a UV-1700 spectrometer (Shimadzu). The total amount of absorbed photons was monitored versusageing time over the whole absorption range for each sample using the ASTM G173 standard as reference for the incident photon flux.39 This allowed for a quantitative comparison of the materials stability. More precisely, the percentage of the light absorbed by the polymer was first calculated for each wavelength using its absorbance spectrum. This number was then multiplied by the amount of incoming photons using the AM1.5G spectrum as a reference for the incoming photonic flux (ASTM G178 standard). This gives the number of absorbed photons at all the wavelengths. The latter was finally summed over the whole polymer absorption band providing the total amount of absorbed photons.Acknowledgements
This work was supported by the Danish Strategic Research Council (DSF 2104-05-0052 and 2104-07-0022)References
- P. L. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. J. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839 CrossRef CAS.
- M. Helgesen, R. Søndergaard and F. C. Krebs, J. Mater. Chem., 2010, 20, 36 RSC.
- F. C. Krebs, T. D. Nielsen, J. Fyenbo, M. Wadstrom and M. S. Pedersen, Energy Environ. Sci., 2010, 3, 512 RSC.
- T. D. Nielsen, C. Cruickshank, S. Foged, J. Thorsen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2010, 94, 1553 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 465 CrossRef CAS.
- F. C. Krebs and K. Norrman, ACS Appl. Mater. Interfaces, 2010, 2, 877 CrossRef CAS.
- F. C. Krebs, T. Tromholt and M. Jørgensen, Nanoscale, 2010, 2, 873 RSC.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 1636 CrossRef CAS.
- F. C. Krebs, Org. Electron., 2009, 10, 761 CrossRef CAS.
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442 RSC.
- F. C. Krebs, J. Fyenbo and M. Jørgensen, J. Mater. Chem., 2010, 20, 8994–9001 RSC.
- http://www.%20solarmer.%20com%2C%20www.%20konarka.%20com/, 2011.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686 CrossRef.
- E. Bundgaard and F. C. Krebs, Macromolecules, 2006, 39, 2823 CrossRef CAS.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. de Boer, Polym. Rev., 2008, 48, 531 Search PubMed.
- E. J. W. List, R. Guentner, P. S. de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374 CrossRef CAS.
- K. L. Chan, M. J. McKiernan, C. R. Towns and A. B. Holmes, J. Am. Chem. Soc., 2005, 127, 7662 CrossRef CAS.
- J. H. Hou, H. Y. Chen, S. Q. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144 CrossRef CAS.
- P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514 CrossRef CAS.
- H. Usta, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 9034 CrossRef CAS.
- E. G. Wang, L. Wang, L. F. Lan, C. Luo, W. L. Zhuang, J. B. Peng and Y. Cao, Appl. Phys. Lett., 2008, 92, 033307 CrossRef.
- J. Y. Wang, S. K. Hau, H. L. Yip, J. A. Davies, K. S. Chen, Y. Zhang, Y. Sun and A. K. Y. Jen, Chem. Mater., 2011, 23, 765 CrossRef CAS.
- H. Y. Chen, J. H. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371 CrossRef CAS.
- M. Morana, H. Azimi, G. Dennler, H. J. Egelhaaf, M. Scharber, K. Forberich, J. Hauch, R. Gaudiana, D. Waller, Z. H. Zhu, K. Hingerl, S. S. van Bavel, J. Loos and C. J. Brabec, Adv. Funct. Mater., 2010, 20, 1180 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS.
- A. J. Moulé, A. Tsami, T. W. Buennagel, M. Forster, N. M. Kronenberg, M. Scharber, M. Koppe, M. Morana, C. J. Brabec, K. Meerholz and U. Scherf, Chem. Mater., 2008, 20, 4045 CrossRef CAS.
- M. Helgesen, S. A. Gevorgyan, F. C. Krebs and R. A. J. Janssen, Chem. Mater., 2009, 21, 4669 CrossRef CAS.
- H. X. Zhou, L. Q. Yang, S. Q. Xiao, S. B. Liu and W. You, Macromolecules, 2010, 43, 811 CrossRef CAS.
- M. Helgesen and F. C. Krebs, Macromolecules, 2010, 43, 1253 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2001 Search PubMed.
- L. J. Lindgren, X. Wang, O. Inganas and M. R. Andersson, Synth. Met., 2005, 154, 97 CrossRef CAS.
- A. Rivaton, B. Mailhot, J. Soulestin, H. Varghese and J. L. Gardette, Eur. Polym. J., 2002, 38, 1349 CrossRef CAS.
- D. M. deLeeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53 CrossRef.
- A. K. Pandey, J. M. Nunzi, B. Ratier and A. Moliton, Phys. Lett. A, 2008, 372, 1333 CrossRef CAS.
- D. Gupta, M. Bag and K. S. Narayan, Appl. Phys. Lett., 2008, 93 Search PubMed.
- F. C. Krebs, K. O. Sylvester-Hvid and M. Jørgensen, Prog. Photovoltaics, 2011, 19, 97–112 CAS.
- NREL's AM1. 5 Standard Dataset, http://rredc.%20nrel.%20gov/solar/spectra/am1.%205/, accessed November, 2010.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures and characterization data including NMR spectra and AFM images of the polymers. See DOI: 10.1039/c1py00017a |
| This journal is © The Royal Society of Chemistry 2011 |