Multi-functionalization of helical block copoly(α-peptide)s by orthogonal chemistry†
Haoyu
Tang
and
Donghui
Zhang
*
Department of Chemistry and Macromolecular Studies Group, Louisiana State University, Baton Rouge, LA, USA. E-mail: dhzhang@lsu.edu; Fax: +1 2255783458; Tel: +1 2255784893
First published on 11th March 2011
Abstract
Diblock copoly(α-peptide)s bearing functional side-chains (i.e., azido or allyl) that are readily derivatized by “click” chemistry have been synthesized by primary amine-initiated sequential polymerization of the corresponding N-carboxyanhydrides (NCAs). Facile derivation of the side-chains has been demonstrated, in which mannose moieties are quantitatively attached via the alkyne–azide [2 + 3] Huisgen cycloaddition to form amphiphilic block copoly(α-peptide)s that retain their helical conformations in solution. Allyl side-chains residing in the hydrophobic core can be further functionalized with high efficiency by radical thiol–ene addition reactions despite the tendency of the amphiphilic precursors to aggregate.
Introduction
Poly(α-peptide)s are attractive biomimetic polymers owing to their ability to fold into well-defined secondary structures (e.g., α-helix and β-sheet)1 and undergo helix-to-coil conformational transitions, a desired attribute for stimuli-responsive materials. Research efforts that focus on the development and application of poly(α-peptide)-based materials have gained much momentum due to the recent advancement in polymerization catalysts that enable access to poly(α-peptide)s with well-defined structures.2–7 As a result of their demonstrated biocompatibility8 and biodegradability,9 poly(α-peptide)-based materials have immense potential in biomedical applications10 such as tissue-engineering scaffolds,11,12drug-delivery carriers,13,14 biomedical imaging agents,15antimicrobial agents,16–18 immunogenic modulators,19,20 and probes for ligand–protein interactions.21–23Poly(α-peptide) or poly(α-peptide)-containing polymers with diverse architectures can be prepared by controlled ring-opening polymerization of N-carboxyanhydrides (NCAs) derived from appropriate amino acids precursors.2–7,24,25 This approach requires the synthesis of individually designed NCA monomers. Additionally, diversity of the side-chain functionality is often limited to prevent interference with the controlled progression of polymerization. For example, poly(L-glutamic acid) and poly(L-lysine) are both synthesized by polymerization of their respective γ-ester or ε-carbamate NCA monomers, affording poly(γ-substituted-L-glutamate) and poly(ε-substituted-L-lysine), in which the side-chains are deprotected post-polymerization. For many biomedical or research applications, covalent attachment of synthetic polymers to biologically active ligands or surfaces is often required. Synthetic strategies that allow for facile and controlled modification of the side-chain structures of poly(α-peptide)s are therefore important for broadening their utility in biomedical fields.
Click chemistry has been extensively used for the modification of biomolecules26–28 and synthetic polymers29–34 due to their high efficiency, specificity, mild reaction conditions and tolerance of functional groups.35,36 The orthogonal chemistry of many click reactions has been utilized for one-pot or sequential functionalization of block and random copolymers, as well as dendrimers, yielding multi-functional materials.34,37–39 For example, Yang and Weck have synthesized poly(norbornene) block copolymers bearing azide and aldehyde side-chains and demonstrated efficient one-pot side-chain functionalization with a variety of molecules having alkyne or hydrazine groups.40 Maynard and coworkers reported the synthesis of reactive block copolymer scaffolds bearing activated ester and protected aldehyde side-chains and their step-wise and selective functionalization with amine and aminooxy molecules.41 Nilles and Theato reported the synthesis of block and random copolymers bearing a single type of reactive side-chain (i.e., pentafluorophenyl ester) that undergoes stepwise and selective reactions with anilines and amines as a result of their different reactivities.42
Synthesis of poly(α-peptide)s with “clickable” side chains by transesterification of poly(γ-benzyl-L-glutamate) has been previously reported.43 However, the efficiency of transesterification reactions is limited. Hammond and coworkers reported the synthesis of poly(γ-propargyl-L-glutamate) by amine-initiated ring-opening polymerizations of the corresponding γ-propargyl-L-glutamic acid-based N-carboxyanhydrides and side-chain grafting of azido-terminated PEO by a copper-mediated [2 + 3] Huisgen cycloaddition.44 Chen and coworkers also demonstrated the grafting of biologically active monosaccharides to the poly(γ-propargyl-L-glutamate) with high efficiency. Tang and Zhang recently reported the quantitative synthesis of complementary poly(γ-azidopropyl-L-glutamate)s by treatment of poly(γ-chloropropyl-L-glutamate)s with NaN3 and the side-chain grafting of mannose moieties.45 Poly(α-peptide)s with allyl or propargyl groups directly attached to α-carbons have also been reported and these side-chains can be further derivatized by thiol–ene addition or alkyne–azide cycloaddition reactions.46,47 While the resulting polypeptides exhibit enhanced hydrolytic stability, they are atactic because racemic NCA monomers are used to prepare the polymers and hence the polymers do not adopt helical conformations. The isotactic analogs have previously been reported by Blanch and coworkers who first resolved the corresponding racemic N-acetyl amino acid precursors by acylase.48
While block copoly(α-peptide)s have been synthesized and shown to self-assemble into various morphologies in solution,49e.g.vesicles,50–55 sheets54 and hydrogels,56–58 their structures are mainly limited to poly(α-peptide)s based on naturally occurring amino acids [e.g., poly(L-glutamic acid), poly(aspartic acid), poly(L-lysine), poly(L-arginine), poly(L-leucine), poly(L-isoleucine)] and a handful of derivatives [e.g., poly(γ-substituted-L-glutamate), PEGylated poly(L-lysine)].52,53 Development of block copoly(α-peptide)s whose side-chains can be orthogonally derivatized with high efficiency will lead to a plethora of structurally diverse multi-functional materials.
In this contribution we report the synthesis and characterization of block copoly(α-peptide)s bearing side-chain functionalities that are amendable to orthogonal “click” chemistry [i.e., poly(γ-allyl-L-glutamate)-b-poly(γ-azidopropyl-L-glutamate) (PALG-b-PAPLG)]. The block copoly(α-peptide)s have been synthesized by primary amine-initiated sequential ring-opening polymerizations of γ-allyl-L-glutamic acid-based NCAs (AL-NCA) and γ-3-chloropropanyl-L-glutamic acid-based NCAs (CP-NCA) followed by nucleophilic substitution with NaN3. The allyl and azido functionalities on the resulting block copoly(α-peptide) side-chains can be readily derivatized by copper-mediated alkyne–azide [2 + 3] cycloaddition or radical thiol–ene addition reactions to confer control over solubility, bio-functionality and solution self-assembly properties. This has been demonstrated by the successful synthesis of amphiphilic poly(γ-mannose-L-glutamate)-b-poly(γ-ally-L-glutamate) block copolymers that retain α-helical conformations. Subsequent grafting of 3-mercaptopropionic acid onto the allyl groups results in the formation of doubly hydrophilic poly(γ-mannose-L-glutamate)-b-poly[γ-(3-mercapto-propanoic acid)-propanyl-L-glutamate] block copolymers.
Experimental section
General considerations
All air or moisture sensitive compounds were handled under a nitrogen atmosphere, either using standard Schlenk-line techniques or in a glovebox. Anhydrous tetrahydrofuran (THF), hexane, dichloromethane and N,N-dimethylformamide (DMF) were obtained by passing through activated alumina columns or molecular sieves under an argon atmosphere (Innovative Technology, Inc.). γ-3-Chloropropanyl-L-glutamic acid-based N-carboxyanhydride 5 (Scheme 2) was synthesized by a literature procedure.45γ-Allyl-L-glutamic acid-based N-carboxyanhydrides 3 (Scheme 1) were synthesized from a modified procedure.59,60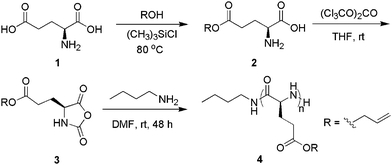 | ||
| Scheme 1 The synthetic route to poly(γ-allyl-L-glutamate) (PALG). | ||
1H and 13C{1H} NMR spectra were recorded on a Bruker AV-400 spectrometer. Chemical shifts are reported in ppm and referenced to the solvent protio impurities and solvent13C{1H} resonances. FTIR spectra were collected on a Bruker Tensor 27 FTIR spectrometer. Size exclusion chromatography (SEC) was performed using an Agilent 1200 system (Agilent 1200 series degasser, isocratic pump, auto sampler and column heater) equipped with three Phenomenex 5 μm, 300 × 7.8 mm columns [100 Å, 1000 Å and Linear (2)], Wyatt DAWN EOS multi-angle light scattering (MALS) detector (GaAs 30 mW laser at λ = 690 nm) and Wyatt Optilab rEX differential refractive index (DRI) detector with a 690 nm light source. DMF containing 0.1 M LiBr was used as the eluent at a flow rate of 0.5 mL min−1. The column temperature was 50 °C and the detector temperature was 25 °C for both MALS and DRI detectors. The refractive index increment (dn/dc) of the synthesized polymers was measured using Wyatt's rEX DRI detector and Astra software dn/dc template. Six polymer/DMF/0.1 M LiBr solutions with different and precise polymer concentrations were sequentially injected into the DRI detector. The measured refractive index values were plotted versus concentration. The slope of a linear fitting of the data is the dn/dc of the polymer. The measured dn/dc of poly(γ-allyl-L-glutamate) (PALG) in DMF/0.1 M LiBr at 25 °C and 690 nm wavelength is 0.0705(18) mL g−1.
CD data were collected with the high tension voltage (i.e., the voltage applied to the photomultiplier) less than 600 V on a Jasco J815 CD spectrometer (Japan Spectroscopic Corporation) with a path length of 0.1 cm and a band width of 1 nm. Two scans were conducted and averaged between 185 nm and 250 nm at a scanning rate of 20 nm min−1 with a resolution of 0.5 nm. The data were processed by subtracting the solvent background and smoothing with Means-Movement method with a convolution of 5. The CD spectra were reported in mean residue ellipticity (MRE) (unit: deg cm2 dmol−1) which was calculated by the equation [θ]λ = MRW × θλ/10 × d × c,61 where MRW is the mean residue weight, θλ is the observed ellipticity (deg) at the wavelength λ, d is the path length and c is the concentration (g mL−1). The copoly(α-peptide) (10.0 mg) was directly dissolved in distilled water using a volumetric flask (10 mL). For temperature-dependent CD studies, the solution was allowed to equilibrate for 5 min prior to data collection. For pH-dependent CD studies, the solution pH was adjusted by the addition of aqueous HCl or KOH.
Dynamic light scattering (DLS) was conducted on a Zetasizer Nano ZS (Malvern Instruments, Ltd., UK) with a He–Ne laser (633 nm) at a scattering angle of 173° (25 °C). Samples (concentration: 1 mg mL−1) were filtered through 0.45 μm membrane filter prior to measurement.
Bright-field images of the micelles were acquired with a JEOL 2011 transmission electron microscope (TEM) operated at 200 kV at −178 °C. The samples were prepared by placing a 10 μL micelle solution (1 mg mL−1) on a holey carbon film grid. The excess solution was absorbed by filter paper. The grid was flash frozen by immediately plunging it into pre-cooled liquid ethane (cooled by liquid nitrogen). The cryo-grid was held in a cryo-holder (Gatan 626) and transferred into the TEM at −178 °C.
Synthesis of γ-allyl-L-glutamate 2
L-Glutamic acid (42.2 g, 0.29 mol) and allyl alcohol (300 mL, 4.42 mol) were mixed in a round-bottom flask (500 mL), followed by addition of chlorotrimethylsilane (40 mL, 0.31 mol) via syringe. The resulting suspension was heated to 80 °C and stirred until it became homogeneous. The solvent was removed at 60 °C under vacuum to give a viscous oil. Addition of diethyl ether (300 mL) to the residue yielded a white solid which was collected by filtration. Additional purification by recrystallization in ethanol/diethyl ether afforded the final product as a white solid (44.9 g, yield: 83%). 1H NMR (D2O, δ, ppm): 5.96 (m, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2-), 5.34 (m, 2H, CH2CHCH2-), 4.65 (d, 2H, CH2CHCH2-), 4.09 (t, 1H, -CHNH2), 2.68 (t, 2H, -COCH2CH2-) and 2.28 (m, 2H, -COCH2CH2-); 13C{1H} NMR (D2O, δ, ppm): 174.57, 172.1, 132.9, 119.20, 66.71, 52.72, 30.23 and 25.53; FTIR (neat, cm−1) 2975, 1738, 1722, 1489, 1223, 1176, 1145, 879 and 836; HR ESI-MS (m/z) [M + H]+ calcd for C8H13NO4, 188.0917; found 188.0904.
CHCH2-), 5.34 (m, 2H, CH2CHCH2-), 4.65 (d, 2H, CH2CHCH2-), 4.09 (t, 1H, -CHNH2), 2.68 (t, 2H, -COCH2CH2-) and 2.28 (m, 2H, -COCH2CH2-); 13C{1H} NMR (D2O, δ, ppm): 174.57, 172.1, 132.9, 119.20, 66.71, 52.72, 30.23 and 25.53; FTIR (neat, cm−1) 2975, 1738, 1722, 1489, 1223, 1176, 1145, 879 and 836; HR ESI-MS (m/z) [M + H]+ calcd for C8H13NO4, 188.0917; found 188.0904.
Synthesis of γ-allyl-L-glutamic acid-based N-carboxyanhydride 3
A round-bottomed flask (250 mL) was charged with γ-allyl-L-glutamate 2 (2.5 g, 13 mmol), triphosgene (1.9 g, 6.4 mmol) and anhydrous THF (40 mL) under nitrogen. The mixture was stirred at room temperature for 24 h over which period the γ-allyl-L-glutamate was gradually dissolved. Removal of the solvent under vacuum yielded an oily liquid which was then dissolved in ethyl acetate (20 mL) and washed with a cold saturated NaHCO3/H2O solution. The organic layer was separated and dried over anhydrous MgSO4 at 0 °C. Filtration and evaporation afforded a clear oil. Recrystallization at −20 °C by layering hexane on top of a CH2Cl2 solution containing the oil was not successful, resulting in two separate liquid layers. The bottom layer was separated and subjected to vacuum to give a clear liquid (1.9 g, yield: 68%). 1H NMR (CDCl3, δ, ppm): 6.52 (s, 1H, NH), 5.91 (m, 1H, CH2CHCH2-), 5.32 (m, 2H, CH2CHCH2-), 4.41 (t, 1H, -CHNH2), 2.59 (t, 2H, -COCH2CH2-) and 2.00–2.40 (m, 2H, -COCH2CH2-); 13C{1H} NMR (CDCl3, δ, ppm): 172.53, 169.72, 152.32, 131.79, 119.34, 66.20, 57.20, 29.94 and 27.15; FTIR (neat, cm−1) 3322, 1852, 1776, 170, 1275, 1173, 1104 and 919; HR ESI-MS (m/z) [M + Na]+ calcd for C9H11NO5, 236.0529; found 236.0513.
Synthesis of poly(γ-allyl-L-glutamate) (PALG) 4
A representative polymerization of 3 is as follows. Inside a glovebox, 3 (146 mg, 0.685 mmol) was dissolved in DMF (5 mL) in a vial. A measured volume of n-butylamine/DMF stock solution (CI = 0.253 M, 136 μL, 34.4 μmol) was subsequently added with syringe. The reaction mixture was stirred at room temperature for 48 h. DMF was distilled off under vacuum at 60 °C. The solid residue was dissolved in CHCl3 and precipitated by cold diethyl ether or methanol. The polymers were collected by centrifugation and decantation and dried under vacuum at room temperature (82 mg, 71% yield). 1H NMR (CDCl3/TFA-d, v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 85:15, δ, ppm): 5.80–6.00 (br m, 1H, CH2CHCH2-), 5.20–5.40 (br m, 2H, CH2CHCH2-), 4.50–4.70 (br m, 3H, CH2CHCH2- and CHNH), 2.40–2.80 (br s, 2H, COCH2CH2-) and 1.90–2.30 (br b, 2H, COCH2CH2-); 13C{1H} NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 175.70, 173.40, 130.69, 119.75, 67.48, 53.58, 30.31 and 27.15.
:v = 85:15, δ, ppm): 5.80–6.00 (br m, 1H, CH2CHCH2-), 5.20–5.40 (br m, 2H, CH2CHCH2-), 4.50–4.70 (br m, 3H, CH2CHCH2- and CHNH), 2.40–2.80 (br s, 2H, COCH2CH2-) and 1.90–2.30 (br b, 2H, COCH2CH2-); 13C{1H} NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 175.70, 173.40, 130.69, 119.75, 67.48, 53.58, 30.31 and 27.15.
Synthesis of poly(γ-allyl-L-glutamate)-b-poly(γ-3-chloropropanyl-L-glutamate) (PALG-b-PCPLG) 6
Inside a glovebox, γ-3-chloropropanyl-L-glutamic acid-based N-carboxyanhydride 5 (172 mg, 0.689 mmol) was added in a vial, followed by adding a measured volume of PALG/DMF solution (C = 0.137 M, 2.0 mL, 0.274 mmol) with syringe. (Note: the PALG/DMF solution used in the block copoly(α-peptide) synthesis was the reaction product directly obtained from the n-butylamine-initiated polymerization of 3 without any purification). The reaction mixture was stirred at room temperature for 48 h and quenched by exposure to air. Removal of the DMF at 40 °C under vacuum yielded a polymer film which was further purified by dissolution in CH2Cl2 (2 mL) and precipitation from methanol (20 mL). Filtration and drying under vacuum at 60 °C for 8 h afforded the final product as a white solid (79 mg, yield of two steps: 42%). 1H NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 5.80–6.00 (br m, 1H, CH2CHCH2-), 5.20–5.40 (br m, 2H, CH2CHCH2-), 4.50–4.70 (br m, 5.3H, CH2CHCH2- and CHNH), 4.20–4.40 (br m, 4.8H, ClCH2CH2CH2-), 3.50–3.70 (br m, 4.7H, ClCH2CH2CH2-), 2.40–2.70 (br s, 6.8H, COCH2CH2-) and 1.90–2.30 (br m, 11.4H, COCH2CH2- and ClCH2CH2CH2-); 13C{1H} NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 175.62, 173.55, 130.77, 119.65, 67.30, 63.39, 53.57, 40.85, 31.09, 30.31 and 27.01.
Synthesis of poly(γ-allyl-L-glutamate)-block-poly(γ-3-azidopropanyl-L-glutamate) (PALG-b-PAPLG) 7
A DMF (5 mL) solution of PALG-b-PCPLG 6 (50 mg, 0.24 mmol of 3-chloropropanyl groups) and sodium azide (0.16 g, 2.4 mmol) was stirred at 60 °C for 48 h and allowed to cool to room temperature. The reaction mixture was filtered to remove any inorganic salts. DMF was removed by vacuum distillation at 60 °C to yield a polymer film which was further purified by dissolution in CH2Cl2, filtration and precipitation in methanol. The resulting polymer was collected by filtration and dried at 60 °C under vacuum (45 mg, yield: 87%). 1H NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 5.80–6.00 (br m, 1H, CH2CHCH2-), 5.20–5.40 (br m, 2H, CH2CHCH2-), 4.50–4.70 (br m, 5.3H, CH2CHCH2- and CHNH), 4.10–4.30 (br m, 4.8H, N3CH2CH2CH2-), 3.30–3.50 (br m, 4.7H, N3CH2CH2CH2-), 2.40–2.70 (br s, 6.8H, COCH2CH2-) and 1.90–2.30 (br m, 11.4H, COCH2CH2- and ClCH2CH2CH2-); 13C{1H} NMR (CDCl3/TFA-d, v:v = 85:15, δ, ppm): 175.78, 173.37, 130.70, 119.70, 67.42, 63.61, 53.57, 48.23, 30.27, 27.59 and 27.19.
Synthesis of poly(γ-allyl-L-glutamate)-block-poly(γ-propanyl-L-glutamate-graft-mannose) [PALG-b-(PPLG-g-mannose)] 8
Inside a glovebox, CuBr (9 mg, 0.063 mmol) was added to a DMF (5 mL) solution of PALG-b-PAPLG 7 (40 mg, 0.19 mmol azido groups), propargylmannose (74 mg, 0.34 mmol) and PMDETA (15 μL, 0.063 mmol). The reaction mixture was stirred at room temperature for 24 h and quenched by exposure to air. Removal of the DMF by vacuum distillation at 60 °C yielded a blue solid which was further purified by dispersion in CH2Cl2/MeOH (4 × 10 mL, v:v = 4:1), centrifugation and decantation to afford a pale yellow solid. The isolated solid was re-dissolved in a saturated EDTA/distilled water solution (100 mL). The solution was dialyzed against distilled water for three days in a dialysis membrane tube with a cutoff molecular weight of 6000–8000 g mol−1. The water was distilled at 60 °C under vacuum to afford a white solid (69 mg, yield: 85%). 1H NMR (D2O, δ, ppm): 8.05 (br s, 1H, CHN3-), 5.86 (br s, 0.7H, CH2CHCH2-), 5.47 (br s, 1.4H, CH2CHCH2-), 4.92 (br s, 1.6H, -CHOCH2-triazole), 4.50–4.70 (br m, 1.4H, -OH), 4.47 (s, 2.8H, CH2CHCH2- and -CHOCH2-triazole), 3.90–4.30 (br m, 3H, -OCH2CH2CH2-triazole and -COCHNH-), 3.50–3.90 (m, 8.2H, mannose and -OCH2CH2CH2-triazole), 1.90–2.90 (br m, 6.7H, -COCH2CH2 and -OCH2CH2CH2-triazole); 13C{1H} NMR (D2O, δ, ppm): 176.32, 174.21, 144.45, 125.47, 133.23, 118.53, 100.01, 73.65, 71.25, 70.68, 67.30, 67.20, 62.41, 61.55, 60.29, 56.96, 47.75, 30.90, 29.38 and 25.75.
Synthesis of 3-mercaptopropionic acid modified poly(γ-allyl-L-glutamate)-block-poly(γ-propanyl-L-glutamate-graft-mannose) [(PALG-g-MCPA)-b-(PPLG-g-mannose)] 9
PALG-b-(PPLG-g-mannose) 8 (20 mg, 19 μmol ally-containing repeating units) and 3-mercaptopropionic acid (MCPA) (17 μL, 190 μmol) were dissolved in degassed distilled water (4 mL) in a 5 mL quartz cell and placed under UV light (254 nm, 16 mW cm−2) for 60 min. The solution was dialyzed against distilled water for three days in a dialysis membrane tube with a cutoff molecular weight of 6000–8000 g mol−1. The water was distilled at 60 °C under vacuum to afford a white solid (15 mg, 70% yield). 1H NMR (D2O, δ, ppm): 8.03 (br s, 1H,CHN3-), 4.91 (br s, 1.2H, -CHOCH2-triazole), 4.50–4.70 (br m, 1.6H, -OH), 4.46 (s, 2.6H, -CHOCH2-triazole), 3.90–4.30 (br m, 4.3H, -OCH2CH2CH2-triazole, -OCH2CH2CH2S- and -COCHNH-), 3.50–3.90 (m, 8.1H, mannose and -OCH2CH2CH2-triazole), 1.90–2.90 (br m, 10.7H, -OCH2CH2CH2SCH2CH2COOH, -COCH2CH2 and -OCH2CH2CH2-triazole); 13C{1H} NMR (D2O, δ, ppm): 177.35, 174.51, 144.15, 125.46, 100.01, 73.58, 71.16, 70.61, 67.29, 62.45, 61.49, 60.30, 56.72, 47.77, 30.85, 29.27 and 25.69.
Results and discussion
Synthesis and characterization of poly(γ-allyl-L-glutamate)s
γ-Allyl-L-glutamic acid-based N-carboxyanhydride (AL-NCA) 3 has been synthesized (Scheme 1) in a good overall yield (50–60%) by a two-step synthetic route based on a modified literature procedure, namely the monesterification of L-glutamic acid with allyl alcohol in the presence of chlorotrimethylsilane (TMSCl)62 and the cyclization of γ-allyl-L-glutamic acid with triphosgene.59,60TMSCl-mediated esterification afforded substantially improved yields of 2 (∼80 to 85%) over those by the literature procedure where sulfuric acid was used to catalyze the reaction (∼25 to 30%).59 The molecular structure of 3 has been verified by 1H NMR, 13C{1H} NMR (Fig. S1†), FTIR spectroscopy (Fig. S2†) and ESI-MS spectrometry.While primary amines have been demonstrated to initiate the polymerization of γ-allyl-L-glutamic acid-based NCAs, control over polymer molecular weight (MW) and molecular weight distribution (PDI) by this method has not been reported. We conducted the polymerization of 3 using n-butylamine initiators under a nitrogen atmosphere in room temperature DMF for 48 h. An aliquot of the reaction solution was used to determine the conversion by FTIR spectroscopy. The polymers were isolated by vacuum distillation of DMF at 60 °C. The polymer structures were determined by 1H NMR, 13C{1H} NMR, FTIR spectroscopy and MALDI-TOF MS spectrometry. The absolute polymer molecular weight (MW) and molecular weight distribution (PDI) were determined by size-exclusion chromatography coupled with multi-angle light scattering and differential refractive index detectors (SEC-MALS-DRI). As the initial monomer to initiator ratio ([3]0:[nBuNH2]0) is increased between 20:1 and 160:1, polymers with MW ranging between 3.0 and 8.3 kg mol−1 and with narrow mono-modal distribution (PDI = 1.04–1.15, Fig. S3†) have been obtained (Table 1). However, the experimental MWs are lower than the theoretical predictions based on a living polymerization with the deviation being more pronounced for polymerizations with larger [3]0:[nBuNH2]0 ratios. We tentatively attribute this to the competitive intramolecular amidation that results in chain termination.
| Entry # | [3]0:[nBu-NH2]0 |
M n (theor.)b/kg·mol−1 | M n (SEC)c/kg·mol−1 | PDIc | Conv.d (%) |
|---|---|---|---|---|---|
|
a
Polymerizations ([3]0 = 0.14 M) were conducted in DMF at room temperature for 48 h using n-butylamine as initiator.
b Theoretical molecular weights were calculated from the [3]0:[nBuNH2]0 ratio and the conversion.
c Absolute polymer molecular weight and molecular weight distribution (PDI) were determined from SEC-MALS-DRI [dn/dc = 0.0705(18) mL g−1, 25 °C, λ = 690 nm].
d Conversions were determined from FTIR spectroscopy.
|
|||||
| 1 | 20 | 3.4 | 3.0 | 1.15 | 100 |
| 2 | 40 | 6.4 | 4.9 | 1.04 | 95 |
| 3 | 80 | 9.9 | 5.9 | 1.05 | 73 |
| 4 | 160 | 15.1 | 8.3 | 1.15 | 56 |
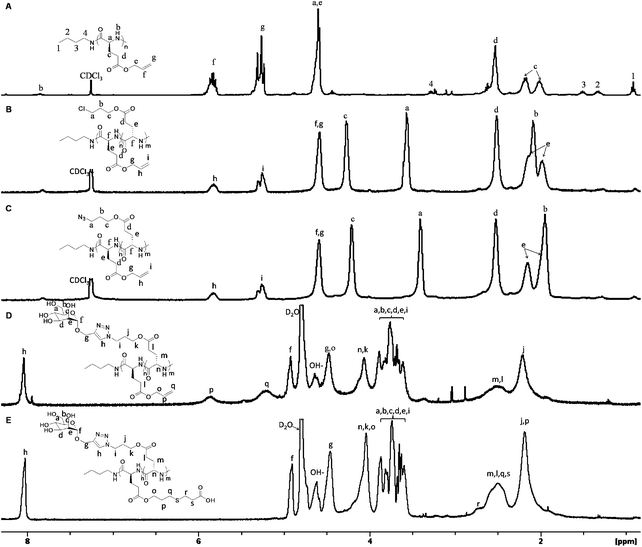
1H (Fig. 1A) and 13C{1H} NMR spectra of the product (Fig. S4†) are consistent with the polymer backbone structure 4 (Scheme 1). MALDI-TOF MS analysis of low MW samples reveals two polymeric species with different end-groups (Fig. 2A and B). The molecular mass of the major species is equal to the sum of an integer number of repeating unit mass (169.07), n-butylamine (73.09) and potassium or sodium ion (38.96 or 22.99) (1 and 2, Fig. 2B). This is consistent with poly(γ-allyl-L-glutamate) (PALG) bearing n-butylamido and amino end groups (1 and 2, Fig. 2C) formed by a primary amine-initiated nucleophilic ring-opening polymerization mechanism.63 Mass analysis of the other minor species is consistent with poly(γ-allyl-L-glutamate) bearing n-butylamido and 5-membered lactam end groups (2, Fig. 2C) formed by intramolecular amidation of the side-chains. The intramolecular amidation competes with chain propagation, resulting in chain termination and lower polymer MWs than theoretical predictions based on living polymerizations. The side-reactions can be minimized by lowering the reaction temperature but at the cost of significantly reduced polymerization rate.64
 | ||
| Fig. 1 (A) 1H NMR spectra of PALG184, (B) PALG18-b-PCPLG366, (C) PALG18-b-PAPLG367 in CDCl3/CF3CO2D (v:v = 85:15), (D) PALG18-b-(PPLG36-g-mannose) 8 and (E) (PALG18-g-MCPA)-b-(PPLG36-g-mannose) 9 in D2O. | ||
 | ||
| Fig. 2 (A) Representative full and (B) expanded MALDI-TOF MS spectra of a low MW PALG (matrix: 3HPA; cation source: CF3CO2K) as well as (C) the molecular structures of PALG with assigned end groups. | ||
Synthesis and characterization of poly(γ-allyl-L-glutamate)-b-poly(γ-3-azidopropanyl-L-glutamate) block copolymer (PALG-b-PAPLG)
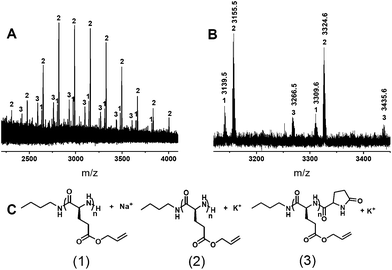
Poly(γ-allyl-L-glutamate)-b-poly(γ-3-chloropropanyl-L-glutamate) block copoly(α-peptide)s (PALG-b-PCPLG) 6 have been synthesized by n-butylamine-initiated sequential polymerization of 3 and 5 (Scheme 2). As intramolecular amidation is more pronounced for the high MW PALG homopolymers, resulting in non-amino chain-ends, low MW PALG 4 (Mn = 3.0 kg mol−1, PDI = 1.15) was targeted and kept invariant in the block copolymer synthesis. PCPLG block length was adjusted to allow for control of block copoly(α-peptide)s composition. SEC chromatography confirms the successful synthesis of the block copoly(α-peptide)s with variable chain length that elute as essentially mono-modal peaks with small shoulders at shorter elution times than the PALG homopolymer precursor (Fig. 3). While the origin of the small high MW shoulders in the SEC chromatograms is not entirely clear, aggregation of the block copolymers under SEC conditions is a likely cause. Alternatively, an “activated monomer” pathway may compete with the primary amine (i.e., PALG 4 macroinitiator)-initiated nucleophilic polymerization of 5, resulting in a small amount of high MW polymer product.651H (Fig. 1B) and 13C{1H} NMR spectroscopy (Fig. S5†) also verifies the formation of the block copoly(α-peptide)s with the desired structure 6 (Scheme 2). Polymer MWs of 6 have been determined by integration of the 1H resonance (a, Fig. 1B) for PCPLG relative to that of PALG (i, Fig. 1B). PALG-b-PCPLG with MW ranging between 10.4 and 30.7 kg mol−1 and reasonably low PDIs (1.08–1.13) can be readily obtained by controlling the [5]0:[nBuNH2]0 ratio. The experimental MWs are in good agreement with theoretical predictions based on living polymerizations (Table 2).
![The synthetic route to 3-mercaptopropionic acid modified poly(γ-allyl-l-glutamate)-block-poly(γ-propanyl-l-glutamate-graft-mannose) [(PALG-g-MCPA)-b-(PPLG-g-mannose)].](/image/article/2011/PY/c1py00015b/c1py00015b-s2.gif) | ||
| Scheme 2 The synthetic route to 3-mercaptopropionic acid modified poly(γ-allyl-L-glutamate)-block-poly(γ-propanyl-L-glutamate-graft-mannose) [(PALG-g-MCPA)-b-(PPLG-g-mannose)]. | ||
 | ||
Fig. 3
SEC
chromatographs of PALG (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), PALG18-b-PCPLG36 ( ), PALG18-b-PCPLG36 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), PALG18-b-PCPLG68 ( ), PALG18-b-PCPLG68 (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) and PALG18-b-PCPLG135 (⋯). ) and PALG18-b-PCPLG135 (⋯). | ||
| Entry | [5]0:[nBuNH2]0 |
M n (theor.)b/kg mol−1 | M n (expt.)c/kg mol−1 | PDId | Conv.e (%) |
|---|---|---|---|---|---|
|
a
Polymerizations ([5]0 = 0.34 M) were conducted in DMF at room temperature for 48 h using PALG (Mn = 3.0 kg mol−1, PDI = 1.15) as initiator.
b Theoretical molecular weight.
c Experimental molecular weight determined from 1H NMR spectroscopy.
d Molecular weight distribution (PDI) was determined from SEC calibrated by PS standards.
e Determined from FTIR spectroscopy by monitoring the disappearance of the CO stretching band (νCO = 1788 cm−1) of 5 in DMF.
|
|||||
| 1 | 50 | 12.8 | 10.4 | 1.08 | 95 |
| 2 | 100 | 22.3 | 17.0 | 1.14 | 94 |
| 3 | 150 | 31.4 | 30.7 | 1.13 | 92 |
Side-chain derivation of poly(γ-allyl-L-glutamate)-b-poly(γ-3-azidopropanyl-L-glutamate) (PALG-b-PAPLG)
Poly(γ-allyl-L-glutamate)-b-poly(γ-3-chloropropanyl-L-glutamate) (PALG18-b-PCPLG36, Mn = 10.4 kg mol−1, PDI = 1.08) 6 was further derivatized into poly(γ-allyl-L-glutamate)-b-poly(γ-3-azidopropanyl-L-glutamate) (PALG18-b-PAPLG36) 7 by treatment with NaN3 in DMF at 60 °C for 48 h (Scheme 2). 1H NMR analysis of the resulting polymers reveals a quantitative conversion, as evidenced by an up-field shift of the methylene proton (a) from 3.57 to 3.40 ppm upon azido substitution (Fig. 1B and C). We have previously demonstrated that the poly(α-peptide) backbones as well ester functional groups on the side-chains remain intact under the substitution reaction conditions.45 Consistently, the integration ratio of vinylene protons (i) of PALG18 to methylene protons (a) of PAPLG36 (i.e., 1:2) exhibits no appreciable change after NaN3 substitution. The presence of the azido group is also evident in the FTIR spectrum of PALG18-b-PAPLG36 (νNNN = 2095 cm−1, Fig. 4D). Quantitative azido substitution has been observed for all three PALG18-b-PCPLGn (n = 36, 68, 135) samples with different PCPLG chain lengths (Fig. S6†).
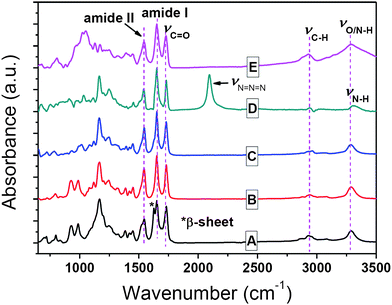 | ||
| Fig. 4 FTIR spectra of (A) PALG18, (B) PALG41, (C) PALG18-b-PCPLG36, (D) PALG18-b-PAPLG36, and (E) PALG18-b-(PPLG36-g-mannose) in the solid state. | ||
The azido and allyl groups on the PALG-b-PAPLG side-chains can be orthogonally derivatized by copper-mediated [2 + 3] alkyne–azide 1,3-dipolar cycloaddition or radical thiol–ene addition chemistry. For example, treatment of PALG18-b-PAPLG36 (Mn = 10.7 kg mol−1, PDI = 1.11) with 1.7 equivalents of mannose derivatives bearing propargyl groups in the presence of CuBr/PMDETA at room temperature for 24 h leads to the formation of the amphiphilic poly(γ-ally-L-glutamate)-b-poly(γ-mannose-L-glutamate) block copolymers [PALG18-b-(PPLG36-g-mannose)] 8 (Scheme 2). While we have previously used 1H NMR analysis to determine the grafting efficiency for PPLG-g-mannose homopolymers,44 this method failed to provide quantitative assessment of the grafting efficiency of mannose due to aggregation of PALG18-b-(PPLG36-g-mannose) in aqueous solutions, resulting in substantial broadening of proton resonances and unreliable integrations (Fig. 1D). FTIR spectroscopy was used instead to verify the grafting efficiency. Complete disappearance of azide vibrational mode has been observed for all three PALG18-b-PCPLGn (n = 36, 68, 135) samples with variable PCPLG chain length, suggesting quantitative grafting of the mannose moieties.
PALG18-b-(PPLGn-g-mannose) (n = 36, 68, 135) block copoly(α-peptide)s exhibit a strong tendency to aggregate in aqueous solution. Cryogenic transmission electron microscopy (CryoTEM) and dynamic light scattering (DLS) studies of PALG18-b-(PPLGn-g-mannose) (n = 36, 68, 135) have confirmed the formation of aggregates with non-uniform size and shape (Fig. S7-A–E†). In spite of their aggregation tendency, the allyl functionalities residing at the hydrophobic core of PALG-b-(PPLG-g-mannose) can be further functionalized by radical thiol–ene addition chemistry. For example, UV irradiation (254 nm, 16 mW cm−2) of PALG18-b-(PPLG36-g-mannose) with ten equivalents of 3-mercaptopropionic acid (MCPA) in distilled water at room temperature for 60 min leads to grafting of the MCPA to the allyl groups to yield the doubly hydrophilic poly[γ-(3-mercaptopropanoic acid)-L-glutamate]-b-poly(γ-mannose-L-glutamate) block copolymer[(PALG18-g-MCPA)-b-(PPLG36-g-mannose)] 9 (Scheme 2 and Fig. 1E). The grafting efficiency is high if not quantitative, as evidenced by the complete disappearance of the 1H resonances due to allyl groups at 5.86 and 5.47 ppm (Fig. 1E). While radical cross-linking between allyl groups is possible, it is not competitive with the desired thiol–ene addition reaction.66
Solid and solution state conformation of block copoly(α-peptide)s
PALGs and their copoly(α-peptide)s have been investigated by FTIR spectroscopy to gain insight to their conformations in the solid state. PALG18 with short chain length (DP = 18) exhibit amide I and amide II bands at 1651 cm−1 and 1545 cm−1 that are characteristic of α-helical conformations. Apart from these bands, another amide I peak characteristic of the β-sheet conformation is notably visible at 1626 cm−1 (Fig. 4A). In comparison, PALG41 with longer chain length (DP = 40) only adopts α-helical conformations in the solid state (Fig. 4B). The chain-length dependent conformations of PALGs are consistent to previous reports for poly(γ-benzyl-L-glutamate)s (PBLGs) where short polypeptides (DP < 18) consist of a mixture of lamellar assembly of β-sheets and columnar hexagonal arrays of α-helices. As the PBLG chain length increases, enhanced cooperative folding results in exclusively α-helical conformations.67 Interestingly, block copoly(α-peptide)s containing short PALG18 blocks [i.e., PALG18-b-PCPLG36, PALG18-b-PAPLG36 and PALG18-b-(PPLG36-g-mannose)] exhibit only α-helical conformations in the solid state, presumably due to enhanced cooperative folding of α-helices and steric incongruity of the side-chains in the block copoly(α-peptide)s that inhibit the efficient packing of β-sheets.The solution conformation of PALG with two different chain lengths (DP = 18 and 41), PALG18-b-PCPLG36, PALG18-b-PAPLG36 and PALG18-b-(PPLG36-g-mannose) has been characterized by CD spectroscopy (Fig. 5). Due to their solubility differences, the CD spectrum for PALG18-b-(PPLG36-g-mannose) was collected in water, whereas the CD spectra of all other samples were measured in THF. PALG18, PALG41, PALG18-b-PCPLG36 and PALG18-b-PAPLG36 adopt α-helical conformations in THF solution, as verified by the characteristic negative ellipticity minima at 208 and 222 nm (Fig. 5). In accord with cooperative folding of poly(α-peptide) α-helices, the longer PALG41 has a much larger mean residual ellipticity (|MRE| = 43 kdeg cm2 dmol−1) than that of the shorter PALG18 (29 kdeg cm2 dmol−1) at 222 nm. While PALG18-b-PCPLG36 and PALG18-b-PAPLG36 block copoly(α-peptide)s also exhibit enhanced MREs relative to that of PALG18, the magnitude of the enhancement differs substantially between the former |MRE| = 43 kdeg cm2 dmol−1 and the latter |MRE| = 33 kdeg cm2 dmol−1 at 222 nm. This suggests that the chain length is not the sole factor that decides the level of cooperativity in α-helix folding for block copoly(α-peptide)s. Steric and chemical compatibility of side-chains is also important in the stability and homogeneity of α-helical conformations.
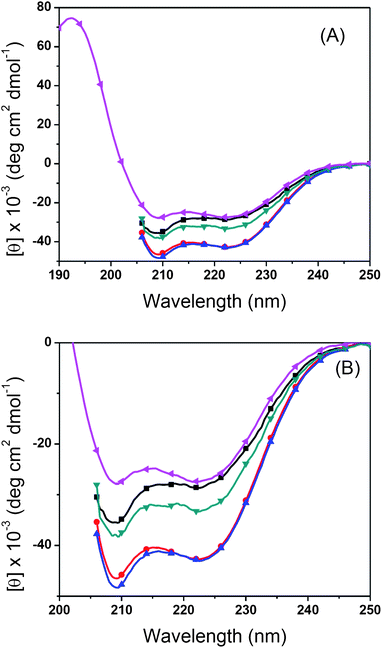 | ||
| Fig. 5 (A) Full and (B) expanded CD spectra of PALG18 (–■–), PALG41 (–●–), PALG18-b-PCPLG36 (–▲–) and PALG18-b-PAPLG36 (–▼–) in THF and PALG18-b-(PPLG36-g-mannose) (–◀–) in water (pH = 7) at 20 °C. | ||
PALG18-b-(PPLG36-g-mannose) also exhibit α-helical conformations in aqueous solution (Fig. 5 and 6). Apart from the negative ellipticity minima at 208 and 222 nm, a characteristic positive ellipticity maximum at 190 nm is also evident. This peak is not observed when CD experiments are conducted in THF due to the absorption of solvent in this wavelength region. The solution conformation of PALG18-b-(PPLG36-g-mannose) has also been investigated at different pHs and temperatures in water. To allow for a quantitative comparison of the relative helical content in PALG18-b-(PPLG36-g-mannose) at different pHs and temperatures, we calculated the fractional helicity (fH) using eqn (1):
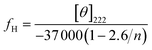 | (1) |
![(A) CD spectra of PALG18-b-(PPLG36-g-mannose) 8 collected at different pHs at 20 °C and (B) at different temperatures in pH = 7 aqueous solutions ([8] = 0.20 mg mL−1).](/image/article/2011/PY/c1py00015b/c1py00015b-f6.gif) | ||
| Fig. 6 (A) CD spectra of PALG18-b-(PPLG36-g-mannose) 8 collected at different pHs at 20 °C and (B) at different temperatures in pH = 7 aqueous solutions ([8] = 0.20 mg mL−1). | ||
As the pH increases from 2 to 12 at 20 °C, the fractional helicity of PALG18-b-(PPLG36-g-mannose) (fH ≈ 80%) remains approximately invariant (Fig. 6A, Table S1: entries 1–5†). A further increase of pH to 13 results in a sharp decrease of fH to 19%, indicating unraveling of the α-helices to coils under strongly basic conditions. The helix-to-coil transition appears to be reversible as the fractional helicity was restored to 65% within 10 min of neutralizing the solution (Table S1: entries 3 and 7†). We attribute the destabilization of α-helical conformations at high pH to electrostatic repulsion of deprotonated mannoses moieties on the side-chains69,70 and partial hydrolytic degradation of ester linkages on the side-chains.
CD analysis of PALG18-b-(PPLG36-g-mannose) at different temperatures reveals the block copoly(α-peptide) conformation is moderately sensitive to temperature variations. As the temperature increases from 5 to 80 °C, the fractional helicity decreases by 35% (Fig. 6B, Table S1: entries 7–11†), suggesting partial uncoiling of the helices. The temperature induced helix-to-coil transition is reversible, as evidenced by a complete recovery of the higher fractional helicity at lower temperatures (Table S1: entries 8–16†).
Conclusions
We have demonstrated the primary-amine initiated polymerization of γ-allyl-L-glutamic acid-based NCAs can yield poly(γ-allyl-L-glutamate)s (PALG) with low to moderate MW (Mn < 10 kg mol−1) and narrow molecular weight distribution (PDI < 1.15). For low MW samples, the polymer MW is consistent with the theoretical prediction for living polymerization. High MW polymer samples cannot be synthesized by this method due to termination by intramolecular amidation. Synthesis of poly(α-allyl-L-glutamate)-b-poly(γ-azidopropanyl-L-glutamate) block copolymers (PALG-b-PAPLG) has been accomplished by polymerization of γ-3-chloropropyl-L-glutamic acid-based NCAs initiated by a short-chain PALG and post-polymerization treatment with NaN3. The azido and allyl functionalities on the resulting block copoly(α-peptide) side-chains can be readily derivatized with quantitative efficiency. This has been demonstrated by the successful synthesis of amphiphilic poly(γ-mannose-L-glutamate)-b-poly(γ-ally-L-glutamate) and doubly hydrophilic poly(γ-mannose-L-glutamate)-b-poly[γ-(3-mercaptopropanoic acid)-propanyl-L-glutamate] block copolymers by copper-mediated alkyne–azide [2 + 3] cycloaddition and radical thiol–ene addition reactions. All block copoly(α-peptide)s adopt α-helical conformations both in solid state and in solution. This methodology provides a convenient and modular approach towards helical block copoly(α-peptides) with diverse structures by side-chain conjugation, allowing control over polymer bioactivity, solubility and self-assembly properties.Acknowledgements
This work is supported by LSU, Louisiana Board of Regents [LEQSF(2008–11)-RD-A-11] and NSF (CHE 0955820). DHZ thanks Dr Jibao He at the Coordinated Instrumentation Facility (CIF) in Tulane University for assisting with the CryoTEM experiments. HYT Thanks Dr Li Guo and Wayne Huberty for assisting with the SEC experiments.References
- H. Block, Poly(γ-Benzyl-L-Glutamate) and Other Glutamic Acid Containing Polymers, Polymer Monographs, Gordon and Breach Science Publisher, New York, 1983, vol. 9 Search PubMed.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2008, 130, 12562–12563 CrossRef CAS.
- H.-A. Klok and T. J. Deming, in Macromolecular Engineering, ed. Y. Gnanou, K. Matyjaszewski and L. Leibler, Wiley, New York, 2007, vol. 1, pp. 519–540 Search PubMed.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- C.-Y. Yang, B. Song, Y. Ao, A. P. Nowak, R. B. Abelowitz, R. A. Korsak, L. A. Havton, T. J. Deming and M. V. Sofroniew, Biomaterials, 2009, 30, 2881–2898 CrossRef CAS.
- M. Obst and A. Steinbüchel, Biomacromolecules, 2004, 5, 1166–1176 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- E. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2008, 19, 109–117 CrossRef CAS.
- C. Zhou, X. Qi, P. Li, W. N. Chen, L. Mouad, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomacromolecules, 2010, 11, 60–67 CrossRef CAS.
- M. Sela and E. Katchalski, Adv. Protein Chem., 1959, 14, 391–478 CrossRef CAS.
- M. D. Wyrsta, A. L. Cogen and T. J. Deming, J. Am. Chem. Soc., 2001, 123, 12919–12920 CrossRef CAS.
- M. B. Bornstein, A. I. Miller, D. Teitelbaum, R. Arnon and M. Sela, Ann. Neurol., 1982, 11, 317–319 CrossRef CAS.
- M. Fridkis-Hareli, E. F. Rosloniec, L. Fugger and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12528–12531 CrossRef CAS.
- N. Ihara, S. Schmitz, M. Kurisawa, J. E. Chung, H. Uyama and S. Kobayashi, Biomacromolecules, 2004, 5, 1633–1636 CrossRef CAS.
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS.
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef CAS.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS.
- K. Aoi, K. Tsutsumiuchi and M. Okada, Macromolecules, 1994, 27, 875–877 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6988 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560–1598 RSC.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS.
- J.-F. Lutz and H. Schlaad, Polymer, 2008, 49, 817–824 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- P. Goyal, K. Yoon and M. Weck, Chem.–Eur. J., 2007, 13, 8801–8810 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- S. K. Yang and M. Weck, Macromolecules, 2007, 41, 346–351.
- R. C. Li, J. Hwang and H. D. Maynard, Chem. Commun., 2007, 3631–3633 RSC.
- K. Nilles and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3683–3692 CrossRef CAS.
- J. Guo, Y. Huang, X. Jing and X. Chen, Polymer, 2009, 50, 2847–2855 CrossRef CAS.
- A. C. Engler, H. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS.
- H. Tang and D. Zhang, Biomacromolecules, 2010, 11, 1585–1592 CrossRef CAS.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef CAS.
- R. M. Guinn, A. O. Margot, J. R. Taylor, M. Schumacher, D. S. Clark and H. W. Blanch, Biopolymers, 1995, 35, 503–512 CrossRef CAS.
- H. Schlaad, Adv. Polym. Sci., 2006, 202, 53–73 CAS.
- J. Rodrí-guez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS.
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS.
- M. Yu, A. P. Nowak, D. P. Pochan and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 12210–12211 CrossRef CAS.
- J. A. Hanson, Z. Li and T. J. Deming, Macromolecules, 2010, 43, 6268–6269 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- E. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- V. Breedveld, A. P. Nowak, J. Sato, T. J. Deming and D. J. Pine, Macromolecules, 2004, 37, 3943–3953 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- D. S. Poché, S. J. Thibodeaux, V. C. Rucker, I. M. Warner and W. H. Daly, Macromolecules, 1997, 30, 8081–8084 CrossRef CAS.
- W. H. Daly and D. S. Poché, Tetrahedron Lett., 1988, 29, 5859–5862 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CAS.
- D. J. Ager, S. Babler, R. A. Erickson, D. E. Froen, J. Kittleson, D. P. Pantaleone, I. Prakash and B. Zhi, Org. Process Res. Dev., 2004, 8, 72–85 Search PubMed.
- T. J. Deming, Adv. Polym. Sci., 2006, 202, 1–18 CAS.
- G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 514–524 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS.
- A. F. Senyurt, H. Wei, C. E. Hayle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901–4909 CrossRef CAS.
- P. Papadopoulos, G. Floudas, H.-A. Klok, I. Schnell and T. Pakula, Biomacromolecules, 2004, 5, 81–91 CrossRef CAS.
- N. A. Besley and J. D. Hirst, J. Am. Chem. Soc., 1999, 121, 9636–9644 CrossRef CAS.
- M. Nagasawa and A. Holtzer, J. Am. Chem. Soc., 1964, 86, 538–543 CrossRef CAS.
- Y. P. Meyer, Macromolecules, 1969, 2, 624–628 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} NMR and FTIR spectra of 3 (Fig. S1 and S2), SEC chromatographs of PALGs (Fig. S3), 1H and 13C{1H} NMR spectra of PALG 4 (Fig. S4) and 13C{1H} NMR spectra of PALG-b-PCPLG 6 (Fig. S5), SEC chromatographs of PALG18-b-PCPLGn (n = 36, 68, 135) and corresponding PALG18-b-PAPLGn (n = 36, 68, 135) (Fig. S6), CryoTEM images and DLS size distribution plot of PALG18-b-(PPLGn-g-mannose), in water (Fig. S7), pH and temperature-dependent CD analysis of PALG18-b-(PPLG36-g-mannose) (Table S1). See DOI: 10.1039/c1py00015b |
| This journal is © The Royal Society of Chemistry 2011 |